
Full-Length Transcriptome Sequencing Provides Insights into Flavonoid Biosynthesis in Fritillaria hupehensis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction, Preparation of PacBio SMRT Library, and Sequencing
2.3. Full-Length Transcriptome Profiling
2.4. Transcriptome Annotation
2.5. Identification of Simple Sequence Repeats and Long Non-Coding RNAs
2.6. Identification of Flavonoid-related Genes and Phylogenetic Analysis
3. Results
3.1. Morphology, Full Transcriptome Sequence, and High-Quality Non-Redundant Sequences
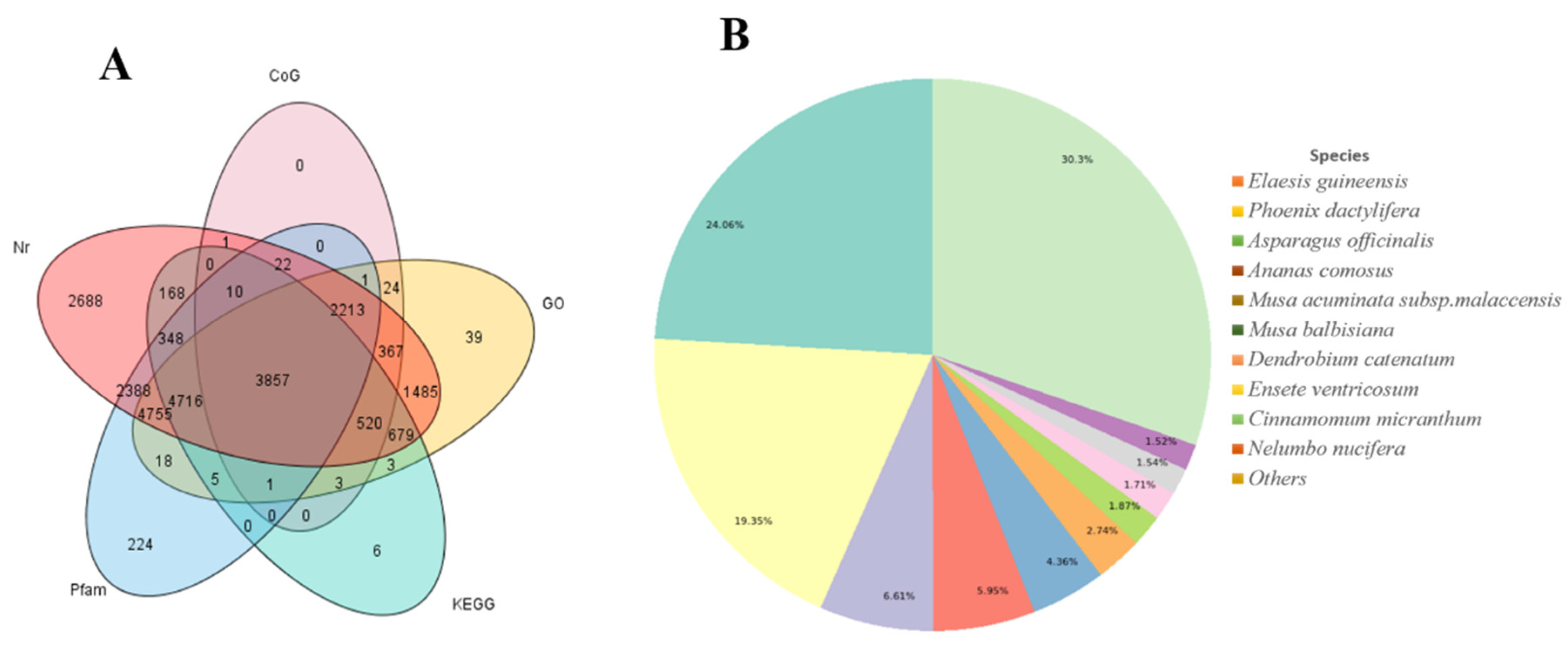
3.2. Functional Annotation of Assembled Transcripts
3.3. Simple Sequence Repeats and Long Non-Coding RNA Analysis
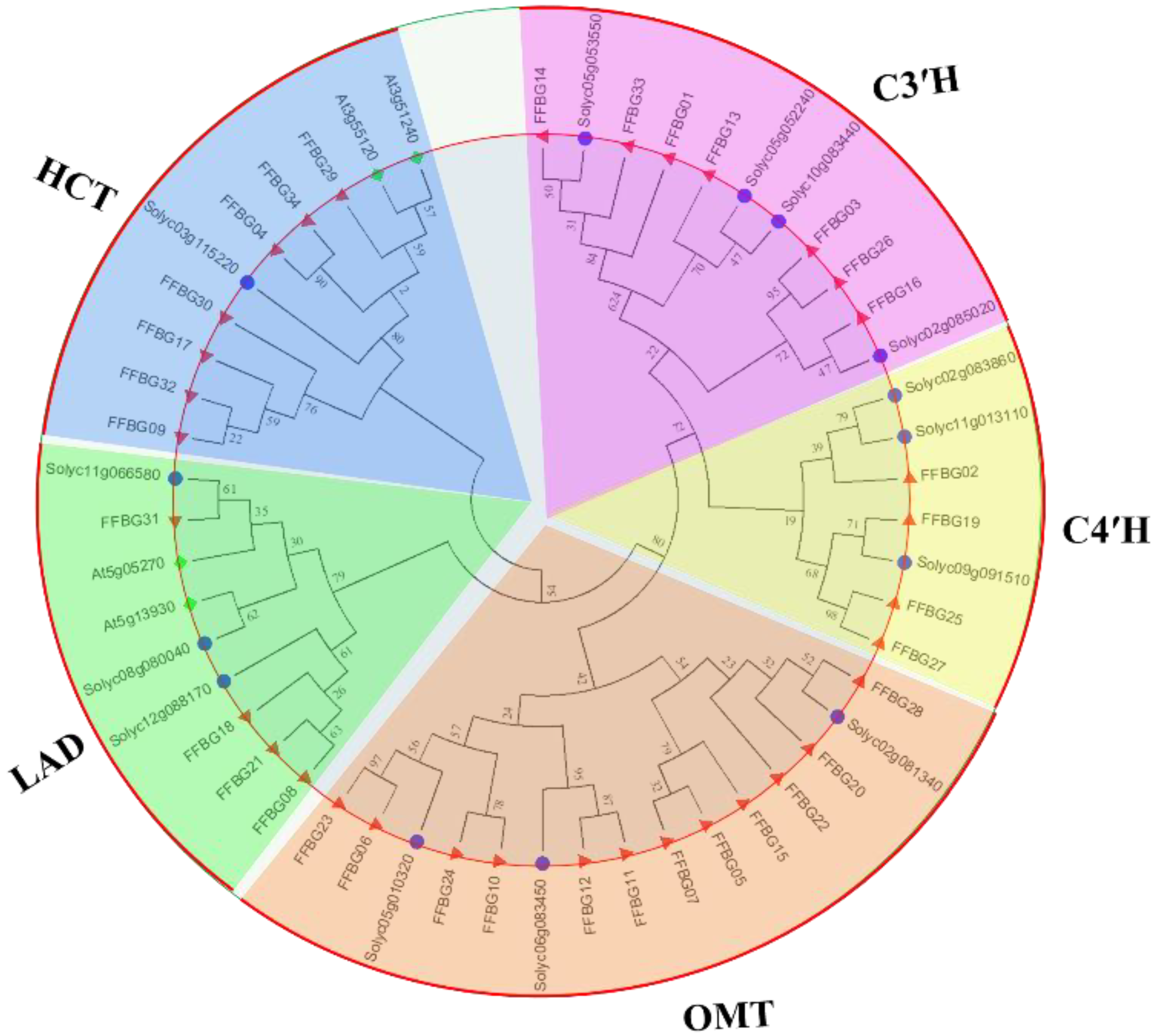
3.4. Candidate Genes involved in Flavonoid Biosynthesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ruan, H.; Zhang, Y.; Wu, J.; Deng, S.; Sun, H.; Fujita, T. Structure of a novel diterpenoid ester, fritillahupehin from bulbs of Fritillaria hupehensis Hsiao and K.C. Hsia. Fitoterapia 2002, 73, 288–291. [Google Scholar] [CrossRef]
- Wang, D.; Wang, S.; Chen, X.; Xu, X.; Zhu, J.; Nie, L.; Long, X. Antitussive, expectorant and anti-inflammatory activities of four alkaloids isolated from Bulbus of Fritillaria wabuensis. J. Ethnopharmacol. 2012, 139, 189–193. [Google Scholar] [CrossRef]
- Pi, H.F.; Zhang, P.; Ruan, H.L.; Zhang, Y.H.; Sun, H.D.; Wu, J.Z. Two new triterpenoids from the leaves and stems of Fritillaria hupehensis. J. Asian Nat. Prod. Res. 2009, 11, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Hao, D.C.; Gu, X.J.; Xiao, P.G.; Peng, Y. Phytochemical and biological research of fritillaria medicine resources. Chin. J. Nat. Med. 2013, 11, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Li, R.; Zhang, Y.; Huang, K.; Wang, W.; Li, J. Transcriptome analysis reveals in vitro-cultured regeneration bulbs as a promising source for targeted Fritillaria cirrhosa steroidal alkaloid biosynthesis. 3 Biotech 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Qiu, F.; Wang, X.; Zheng, Y.; Wang, H.; Liu, X.; Su, X. Full-length transcriptome sequencing and different chemotype expression profile analysis of genes related to monoterpenoid biosynthesis in Cinnamomum porrectum. Int. J. Mol. Sci. 2019, 20, 6230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Partap, M.; Rana, D.; Kumar, P.; Warghat, A.R. Metabolite and expression profiling of steroidal alkaloids in wild tissues compared to bulb derived in vitro cultures of Fritillaria roylei—High value critically endangered Himalayan medicinal herb. Ind. Crops Prod. 2020, 145, 111945. [Google Scholar] [CrossRef]
- Pi, H.F.; Zhang, P.; Zhu, T.; Ruan, H.L.; Zhang, Y.H.; Sun, H.D.; Wu, J.Z. A new cycloartane triterpenoid from the leaves and stems of Fritillaria hupehensis. Chin. Chem. Lett. 2007, 18, 418–420. [Google Scholar] [CrossRef]
- Shujun, W.; Jinglin, Y.; Wenyuan, G.; Jiping, P.; Jiugao, Y.; Peigen, X. Characterization of starch isolated from Fritillaria traditional Chinese medicine (TCM). J. Food Eng. 2007, 80, 727–734. [Google Scholar] [CrossRef]
- Wang, X.; Luo, H.; Wei, X.; Cao, P.; Gao, Z.; Han, J. Transcriptome analysis provides insights into key gene(s) involved in steroidal alkaloid biosynthesis in the medicinally important herb Fritillaria taipaiensis. Res. Sq. 2019, 1–35. [Google Scholar] [CrossRef] [Green Version]
- Xin, J.; Zhang, R.C.; Wang, L.; Zhang, Y.Q. Researches on Transcriptome Sequencing in the Study of Traditional Chinese Medicine. Evid. Based Complement. Altern. Med. 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Sun, C.; Luo, H.M.; Li, X.W.; Niu, Y.Y.; Chen, S.L. Transcriptome characterization for Salvia miltiorrhiza using 454 GS FLX. Yaoxue Xuebao 2010, 45, 524–529. [Google Scholar]
- Sun, C.; Li, Y.; Wu, Q.; Luo, H.; Sun, Y.; Song, J.; Lui, E.M.K.; Chen, S. De novo sequencing and analysis of the American ginseng root transcriptome using a GS FLX Titanium platform to discover putative genes involved in ginsenoside biosynthesis. BMC Genom. 2010, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Song, J.; Sun, Y.; Suo, F.; Li, C.; Luo, H.; Liu, Y.; Li, Y.; Zhang, X.; Yao, H.; et al. Transcript profiles of Panax quinquefolius from flower, leaf and root bring new insights into genes related to ginsenosides biosynthesis and transcriptional regulation. Physiol. Plant. 2010, 138, 134–149. [Google Scholar] [CrossRef]
- Li, Y.; Luo, H.M.; Sun, C.; Song, J.Y.; Sun, Y.Z.; Wu, Q.; Wang, N.; Yao, H.; Steinmetz, A.; Chen, S.L. EST analysis reveals putative genes involved in glycyrrhizin biosynthesis. BMC Genom. 2010, 11, 268. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Zuo, L.; Zuo, T.; Zhang, E.; Luo, X.; Fan, L.; Feng, L. Total saponins contents and anti-inflammatory effect of Fritillariae cirrhosae bulbus of different species. J. Southwest Univ. Natl. 2012, 38, 252–255. [Google Scholar]
- Wang, D.; Yang, J.; Du, Q.; Li, H.; Wang, S. The total alkaloid fraction of bulbs of Fritillaria cirrhosa displays anti-inflammatory activity and attenuates acute lung injury. J. Ethnopharmacol. 2016, 193, 150–158. [Google Scholar] [CrossRef]
- Guo, S. Mining and identification of genes involved in dendrobivun alkaloid and ginsenoslde biosynthesis based on transcriptome analysis. Peking Union Med. Coll. 2013, 10, 21–34. [Google Scholar]
- Gai, Q.Y.; Jiao, J.; Luo, M.; Wang, W.; Gu, C.B.; Fu, Y.J.; Ma, W. Tremendous enhancements of isoflavonoid biosynthesis, associated gene expression and antioxidant capacity in Astragalus membranaceus hairy root cultures elicited by methyl jasmonate. Process Biochem. 2016, 51, 642–649. [Google Scholar] [CrossRef]
- Gao, R.; Hu, Y.; Dan, Y.; Hao, L.; Liu, X.; Song, J. Chinese herbal medicine resources: Where we stand. Chinese Herb. Med. 2020, 12, 3–13. [Google Scholar] [CrossRef]
- Yang, Z.; An, W.; Liu, S.; Huang, Y.; Xie, C.; Huang, S.; Zheng, X. Mining of candidate genes involved in the biosynthesis of dextrorotatory borneol in Cinnamomum burmannii by transcriptomic analysis on three chemotypes. PeerJ 2020, 8, e9311. [Google Scholar] [CrossRef]
- Kotwal, S.; Kaul, S.; Sharma, P.; Gupta, M.; Shankar, R.; Jain, M.; Dhar, M.K. De novo transcriptome analysis of medicinally important plantago ovata using RNA-seq. PLoS ONE 2016, 11, e0150273. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tang, X.; Ren, C.; Wei, B.; Wu, Y.; Wu, Q.; Pei, J. Full-length transcriptome sequences and the identification of putative genes for flavonoid biosynthesis in safflower. BMC Genom. 2018, 19, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, F.; Huang, L.; Qi, L.; Ma, Y.; Yan, Z. Full-length transcriptome analysis of Coptis deltoidea and identification of putative genes involved in benzylisoquinoline alkaloids biosynthesis based on combined sequencing platforms. Plant Mol. Biol. 2020, 102, 477–499. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, I.; Pal, T.; Sood, H.; Chauhan, R.S. Comparative transcriptome analysis in different tissues of a medicinal herb, Picrorhiza kurroa pinpoints transcription factors regulating picrosides biosynthesis. Mol. Biol. Rep. 2016, 43, 1395–1409. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Guo, F.; Zhan, L.; Mohr, T.; Cheng, P.; Huo, N.; Gu, R.; Pei, D.; Sun, J.; et al. Deep sequencing and transcriptome analyses to identify genes involved in secoiridoid biosynthesis in the Tibetan medicinal plant Swertia mussotii. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Zhou, S.; Zhang, J.; Han, H.; Zhang, J.; Ma, H.; Zhang, Z.; Lu, Y.; Liu, W.; Yang, X.; Li, X.; et al. Full-length transcriptome sequences of Agropyron cristatum facilitate the prediction of putative genes for thousand-grain weight in a wheat-A. cristatum translocation line. BMC Genom. 2019, 20, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Zhao, H.X.; Li, P.P.; Zeng, W.J.; Li, Y.H.; Ge, F.W.; Zhao, J.J.; Zhao, H.P. De novo characterization of the seed transcriptome of Lepidium apetalum willd. China Biotechnol. 2016, 36, 38–46. [Google Scholar]
- Lulin, H.; Xiao, Y.; Pei, S.; Wen, T.; Shangqin, H. The first Illumina-based De Novo transcriptome sequencing and analysis of Safflower flowers. PLoS ONE 2012, 7, e38653. [Google Scholar] [CrossRef]
- Li, Q.; Ding, G.; Li, B.; Guo, S.X. Transcriptome analysis of genes involved in dendrobine biosynthesis in dendrobium nobile Lindl infected with mycorrhizal fungus MF23 (Mycena sp.). Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Guo, H.; Chen, H.; Shi, Y.; Meng, Y.; Lu, J.; Feng, S.; Wang, H. Identification and analysis of genes associated with the synthesis of bioactive constituents in Dendrobium officinale using RNA-Seq. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Teranishi, Y.; Ueda, S.; Suzuki, H.; Kawano, N.; Yoshimatsu, K.; Saito, K.; Kawahara, N.; Muranaka, T.; Seki, H. Cytochrome P450 monooxygenase CYP716A141 is a unique β-amyrin C-16β oxidase involved in triterpenoid saponin biosynthesis in Platycodon grandiflorus. Plant Cell Physiol. 2017, 58, 874–884. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, C.; Zhou, C.; Li, J.; Zhang, Y. Molecular characterization of the C -glucosylation for puerarin biosynthesis in Pueraria lobata. Plant J. 2017, 90, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serres-Giardi, L.; Belkhir, K.; David, J.; Glémin, S. Patterns and evolution of nucleotide landscapes in seed plants. Plant Cell 2012, 24, 1379–1397. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Kalderimis, A.; Stepan, R.; Sullivan, J.; Lyne, R.; Lyne, M.; Micklem, G. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. F1000Research 2014, 25, 25–29. [Google Scholar]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 32, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7–R35. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated nr database in protein annotation system and its localization. Nat. Commun. 2010, 32, 71–72. [Google Scholar]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Deng, X.; Liu, Y.; Luo, A.; Chen, J.; Xiang, J.; Zhao, Z. Genetic separation of chalkiness by hybrid rice of Huanghuazhan and CS197. Biocell 2020, 44, 451–459. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [Green Version]
- Neeraja, C.N.; Maghirang-Rodriguez, R.; Pamplona, A.; Heuer, S.; Collard, B.C.Y.; Septiningsih, E.M.; Vergara, G.; Sanchez, D.; Xu, K.; Ismail, A.M.; et al. A marker-assisted backcross approach for developing submergence-tolerant rice cultivars. Theor. Appl. Genet. 2007, 115, 767–776. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Harr, B.; Schlötterer, C. Long microsatellite alleles in Drosaphila melanogaster have a downward mutation bias and short persistence times, which cause their genome-wide underrepresentation. Genetics 2000, 155, 1213–1220. [Google Scholar]
- Li, H.J.; Jiang, Y.; Li, P. Chemistry, bioactivity and geographical diversity of steroidal alkaloids from the Liliaceae family. Nat. Prod. Rep. 2006, 23, 735–752. [Google Scholar] [CrossRef]
- Liu, J.; Peng, C.; He, C.J.; Liu, J.L.; He, Y.C.; Guo, L.; Zhou, Q.M.; Yang, H.; Xiong, L. New amino butenolides from the bulbs of Fritillaria unibracteata. Fitoterapia 2014, 98, 53–58. [Google Scholar] [CrossRef]
- Wong, M.M.L.; Cannon, C.H.; Wickneswari, R. Identification of lignin genes and regulatory sequences involved in secondary cell wall formation in Acacia auriculiformis and Acacia mangium via de novo transcriptome sequencing. BMC Genomics 2011, 12, 342. [Google Scholar] [CrossRef] [Green Version]
- Mizrachi, E.; Hefer, C.A.; Ranik, M.; Joubert, F.; Myburg, A.A. De novo assembled expressed gene catalog of a fast-growing Eucalyptus tree produced by Illumina mRNA-Seq. BMC Genomics 2010, 11, 681. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.K.; Balyan, H.S.; Sharma, P.C.; Ramesh, B. Microsatellites in plants: A new class of molecular markers. Curr. Sci. 1996, 70, 45–54. [Google Scholar]
- Jia, B.; Lin, Q.; Zhang, L.; Tan, X.; Lei, X.; Hu, X.; Shao, F. Development of 15 Genic-SSR markers in oil-tea tree (camellia oleifera) based on transcriptome sequencing. Genetika 2014, 23, 48–55. [Google Scholar] [CrossRef]
- Eddy, S.R. Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2001, 482, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Jalali, S.; Kapoor, S.; Sivadas, A.; Bhartiya, D.; Scaria, V. Computational approaches towards understanding human long non-coding RNA biology. Bioinformatics 2015, 3, 759–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanowski, A.; Yanovsky, M.J. Circadian rhythms and post-transcriptional regulation in higher plants. Front. Plant Sci. 2015, 6, 437–448. [Google Scholar] [CrossRef] [Green Version]
- Beltran, M.; Puig, I.; Peña, C.; García, J.M.; Álvarez, A.B.; Peña, R.; Bonilla, F.; De Herreros, A.G. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev. 2008, 22, 756–769. [Google Scholar] [CrossRef] [Green Version]
- Schuenemann, D.; Gupta, S.; Persello-Cartieauxi, F.; Klimyuk, V.I.; Jones, J.D.G.; Nussaume, L.; Hoffman, N.E. A novel signal recognition particle targets light-harvesting proteins to the thylakoid membranes. Proc. Natl. Acad. Sci. USA 1998, 95, 10312–10316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orfila, C.; Sørensen, S.O.; Harholt, J.; Geshi, N.; Crombie, H.; Truong, H.N.; Reid, J.S.G.; Knox, J.P.; Scheller, H.V. QUASIMODO1 is expressed in vascular tissue of Arabidopsis thaliana inflorescence stems, and affects homogalacturonan and xylan biosynthesis. Planta 2005, 222, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Hichri, I.; Barrieu, F.; Bogs, J.; Kappel, C.; Delrot, S.; Lauvergeat, V. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 2011, 62, 2465–2483. [Google Scholar] [CrossRef] [Green Version]
- Pospiech, A.; Bietenhader, J.; Schupp, T. Two multifunctional peptide synthetases and an O-methyltransferase are involved in the biosynthesis of the DNA-binding antibiotic and antitumour agent saframycin Mx1 from Myxococcus xanthus. Microbiology 1996, 142, 741–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wils, C.R.; Brandt, W.; Manke, K.; Vogt, T. A single amino acid determines position specificity of an Arabidopsis thaliana CCoAOMT-like O-methyltransferase. FEBS Lett. 2013, 587, 683–689. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Reinhard, K.; Schiltz, E.; Matern, U. Characterization and heterologous expression of hydroxycinnamoyl/benzoyl-coA:anthranilate N-hydroxycinnamoyl/benzoyltransferase from elicited cell cultures of carnation, Dianthus caryophyllus L. Plant Mol. Biol. 1997, 35, 777–789. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, B.; Laflamme, P.; Alarco, A.M.; De Luca, V. The terminal O-acetyltransferase involved in vindoline biosynthesis defines a new class of proteins responsible for coenzyme A-dependent acyl transfer. Plant J. 1998, 14, 703–713. [Google Scholar] [CrossRef]
- Liu, X.; Dong, Y.; Yao, N.; Zhang, Y.; Wang, N.; Cui, X.; Li, X.; Wang, Y.; Wang, F.; Yang, J.; et al. De novo sequencing and analysis of the safflower transcriptome to discover putative genes associated with safflor yellow in Carthamus tinctorius L. Int. J. Mol. Sci. 2015, 16, 25657–25677. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Library | Number of Reads | Number of Subreads | Number of FL Transcripts | Number of FLNC | Assembly Length (Mb) | Average Transcript Length (bp) | N50 (bp) |
|---|---|---|---|---|---|---|---|
| Reads | 342,044 | 28,880,638 | 316,438 | 274,919 | 1647 | 1365 | 1888 |
| SSR | Number of SSR |
|---|---|
| Total SSRs | 7914 |
| Total SSR length | 13387 |
| Relative abundance (SSR/Mb) | 143 |
| Relative density (bp/Mb) | 243 |
| SSR containing sequences | 5973 |
| Sequences containing more than 1 SSR | 1311 |
| LncRNA Length | Number |
|---|---|
| 200–400 | 1899 |
| 400–600 | 5279 |
| 600–800 | 7986 |
| 800–1000 | 7180 |
| 1000–1200 | 5058 |
| 1200–1400 | 3382 |
| 1400–1600 | 2113 |
| 1600–1800 | 1438 |
| 1800–2000 | 909 |
| 2000–2200 | 723 |
| 2200–2400 | 529 |
| 2400–2600 | 391 |
| 2600–2800 | 314 |
| 2800–3000 | 270 |
| 3000–3200 | 221 |
| 3200–3400 | 185 |
| 3400–3600 | 140 |
| 3600–3800 | 118 |
| 3800–4000 | 96 |
| 4000–4200 | 70 |
| 4200–4400 | 78 |
| 4400–4600 | 49 |
| 4600–4800 | 39 |
| 4800–5000 | 27 |
| 5000–5200 | 26 |
| 5200–5400 | 13 |
| 5400–5600 | 20 |
| 5600–5800 | 15 |
| 5800–6000 | 8 |
| >6000 | 31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, K.; Chen, J.; Niu, Y.; Lin, X. Full-Length Transcriptome Sequencing Provides Insights into Flavonoid Biosynthesis in Fritillaria hupehensis. Life 2021, 11, 287. https://doi.org/10.3390/life11040287
Guo K, Chen J, Niu Y, Lin X. Full-Length Transcriptome Sequencing Provides Insights into Flavonoid Biosynthesis in Fritillaria hupehensis. Life. 2021; 11(4):287. https://doi.org/10.3390/life11040287
Chicago/Turabian StyleGuo, Kunyuan, Jie Chen, Yan Niu, and Xianming Lin. 2021. "Full-Length Transcriptome Sequencing Provides Insights into Flavonoid Biosynthesis in Fritillaria hupehensis" Life 11, no. 4: 287. https://doi.org/10.3390/life11040287
APA StyleGuo, K., Chen, J., Niu, Y., & Lin, X. (2021). Full-Length Transcriptome Sequencing Provides Insights into Flavonoid Biosynthesis in Fritillaria hupehensis. Life, 11(4), 287. https://doi.org/10.3390/life11040287