
Transcriptome Profiling Reveals Molecular Changes during Flower Development between Male Sterile and Fertile Chinese Cabbage (Brassica rapa ssp. pekinensis) Lines

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Growth Conditions
2.2. RNA Extraction and Illumina Sequencing
2.3. Analysis of the RNA-Seq Data
2.4. Functional Classification and Pathway Enrichment Analysis
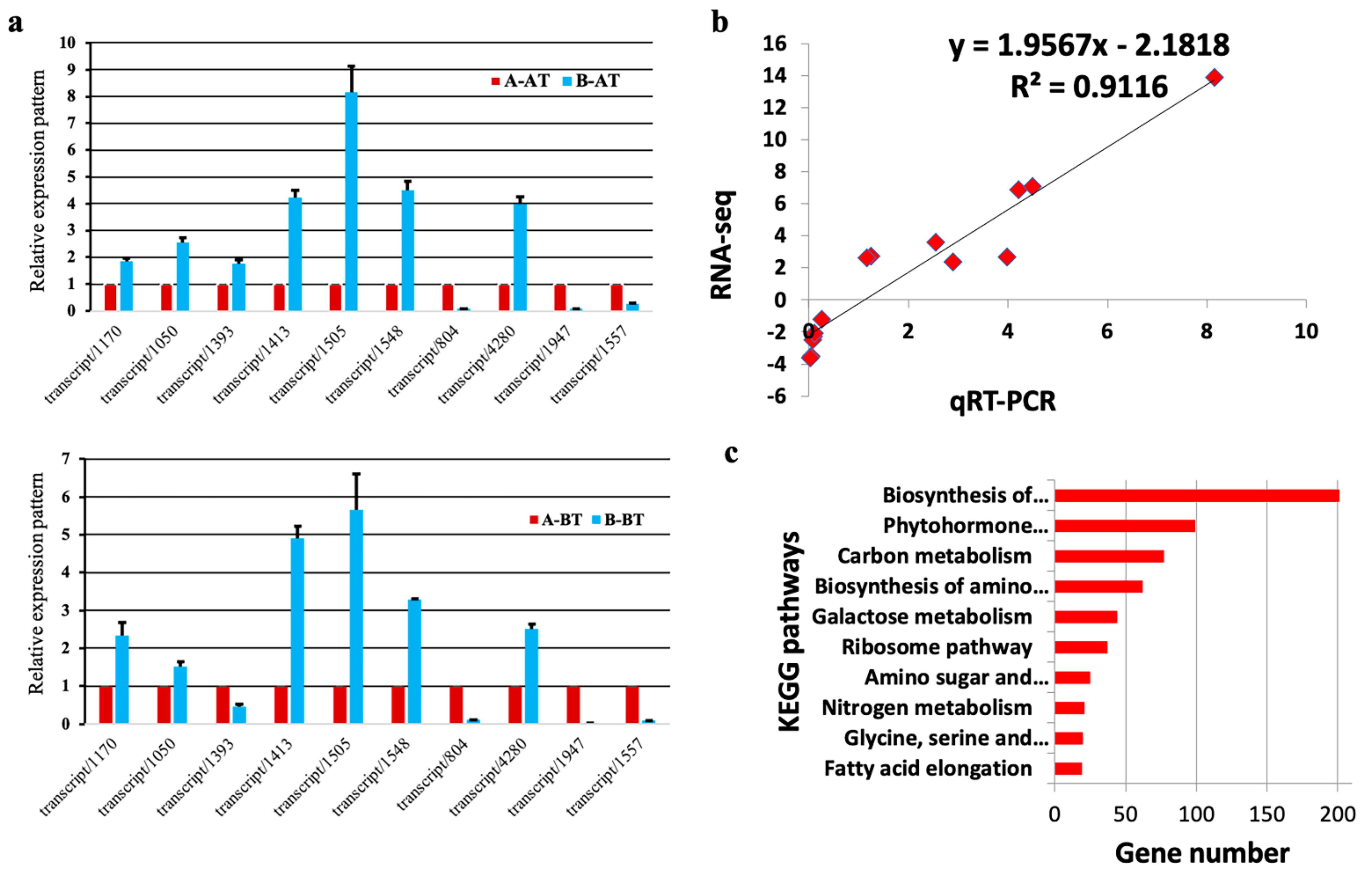
2.5. RNA-Seq Data Evaluation
3. Results
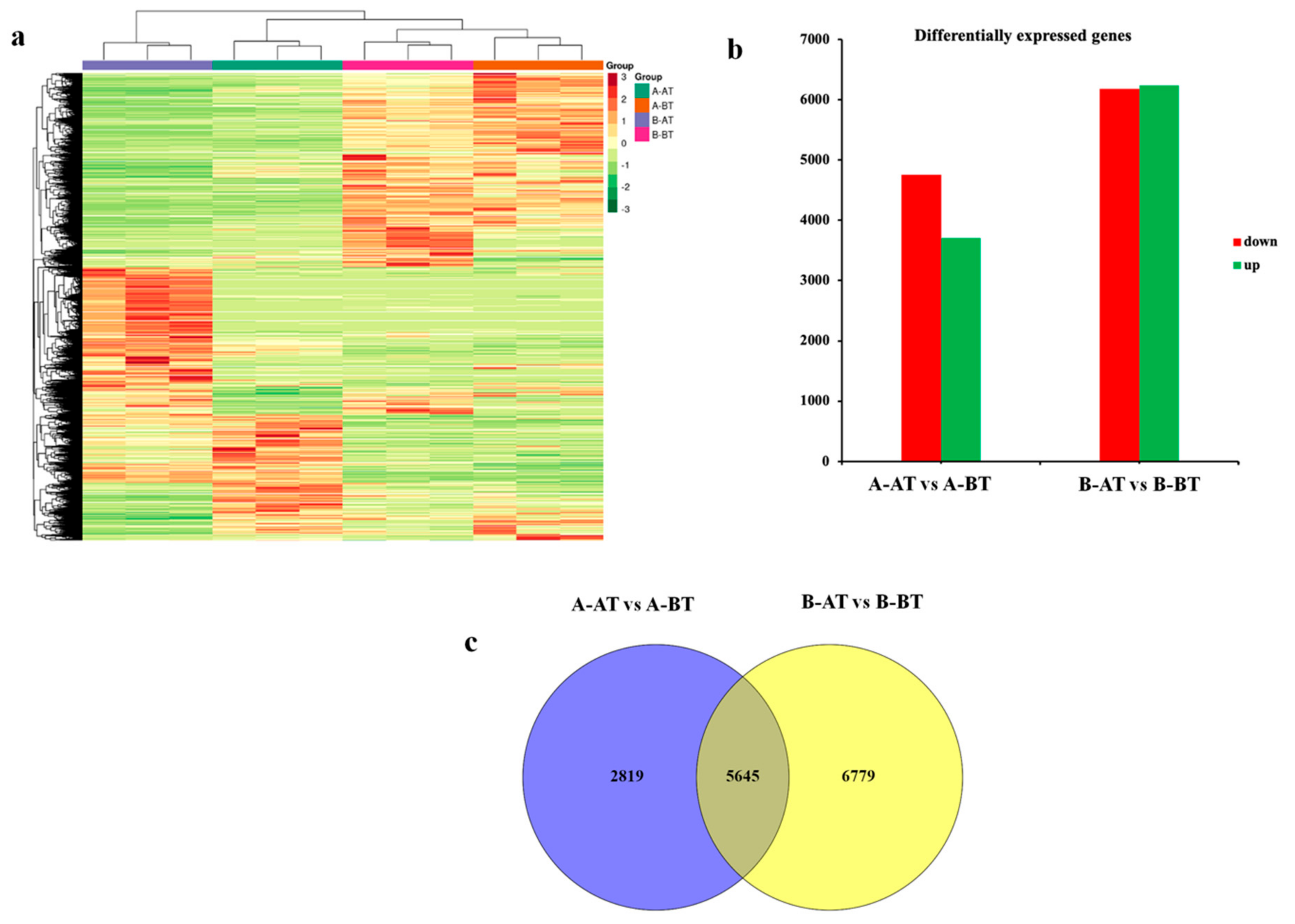
3.1. Overview of the RNA-Seq and Differential Genes Expression Analyses
3.2. Differentially Expressed Transcription Factors
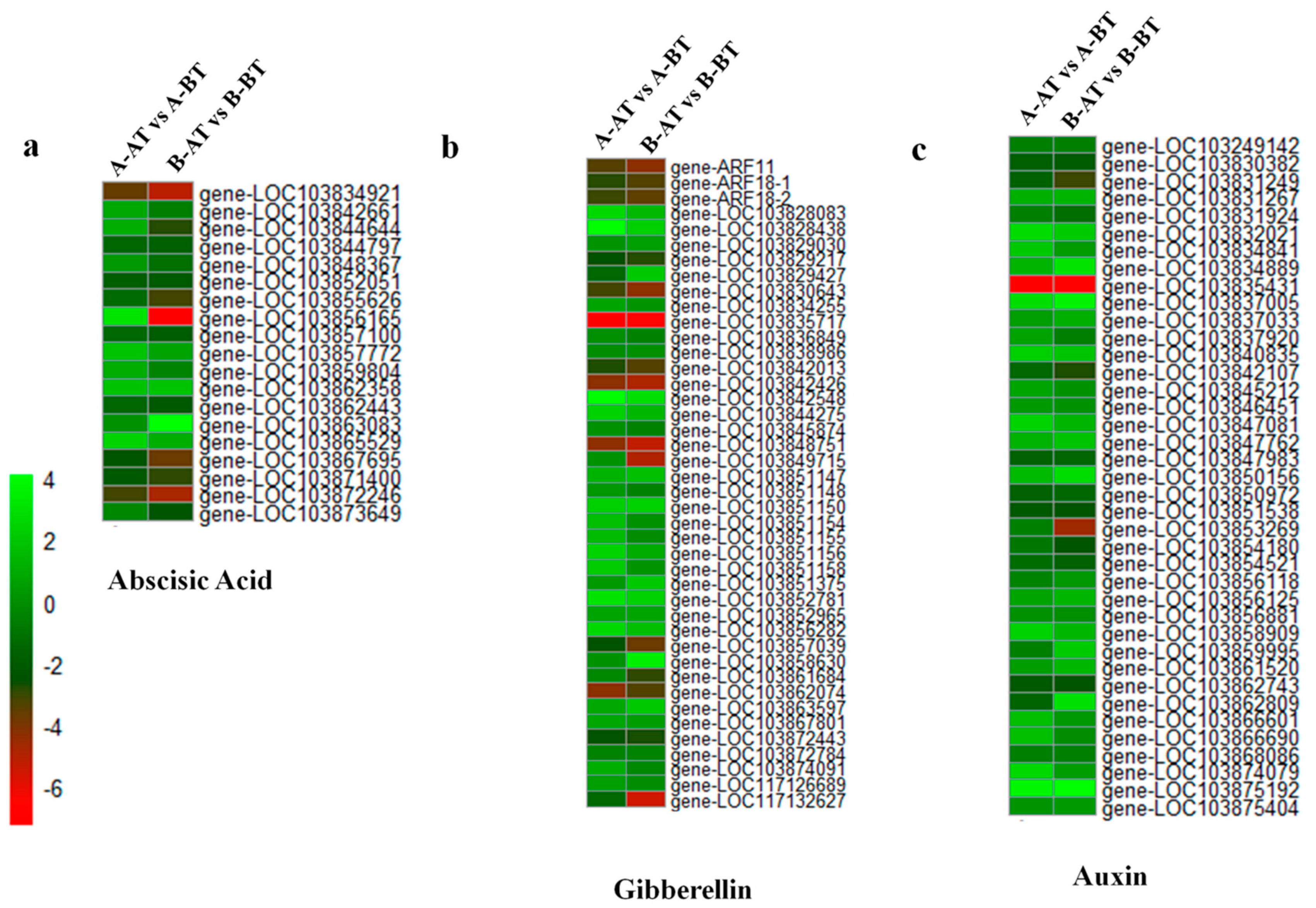
3.3. Genes Related to Phytohormones
3.4. DEGs Involved in Carbon Metabolism
3.5. DEGs Related to the Biosynthesis of Amino Acids
3.6. Genes Related to Anther and Pollen Development
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saxena, K.B.; Hingane, A.J. Male sterility systems in major field crops and their potential role in crop improvement. Plant Biol. Biotechnol. 2015, 1, 639–656. [Google Scholar]
- Hui, F.; Ning, Y.; Zhiyong, L.; Hao, W. A genetic male sterile line developed by molecular marker-assisted selection in Chinese cabbage (Brassica rapa ssp. pekinensis). Afr. J. Biotechnol. 2011, 10, 17706–17711. [Google Scholar]
- Qu, C.; Fu, F.; Liu, M.; Zhao, H.; Liu, C.; Li, J.; Tang, Z.; Xu, X.; Qiu, X.; Wang, R.; et al. Comparative Transcriptome Analysis of Recessive Male Sterility (RGMS) in Sterile and Fertile Brassica napus Lines. PLoS ONE 2015, 10, e0144118. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Sun, C.; Li, H.; Hu, S.; Lei, L.; Kang, J. Integrated analysis of transcriptome and proteome changes related to the ogura cytoplasmic male sterility in cabbage. PLoS ONE 2018, 13, e0193462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Z.; Liu, Y.; Lou, P.; Liu, G. Current trends in cabbage breeding. J. New Seeds 2004, 6, 75–107. [Google Scholar] [CrossRef]
- Murai, K.; Takumi, S.; Koga, H.; Ogihara, Y. Pistillody, homeotic transformation of stamens into pistil-like structures, caused by nuclear-cytoplasm interaction in wheat. Plant J. 2002, 29, 169–181. [Google Scholar] [CrossRef]
- Tang, J.J.; Chen, X.; Hu, Q.D.; Kato, M.; Shimizu, K.; Yokoo, M. A comparatively histological observation on the megagametophytic abortion of female-sterile rice FS-1 and its maternal parent Fujisaka 5. Shi Yan Sheng Wu Xue Bao 2002, 35, 313–318. [Google Scholar]
- Jin, W.; Palmer, R.G.; Horner, H.T.; Shoemaker, R.C. Molecular mapping of a male-sterile gene in soybean. Crop Sci. 1998, 38, 1681–1685. [Google Scholar] [CrossRef] [Green Version]
- Arthur, L.; Ozias-akins, P.; Hanna, W.W. Female sterile mutant in pearl millet: Evidence for initiation of apospory. J. Hered. 1993, 84, 112–115. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, G.; Primard, C.; Vedel, F.; Chetrit, P.; Remy, R.; Rousselle, R.M. Intergeneric cytoplasmic hybridization in cruciferae by protoplast fusion. MGG Mol. Gen. Genet. 1983, 191, 244–250. [Google Scholar] [CrossRef]
- Chase, C.D. Cytoplasmic male sterility: A window to the world of plant mitochondrial-nuclear interactions. Trends Genet. 2007, 23, 81–90. [Google Scholar] [CrossRef]
- Dong, X.; Kim, W.K.; Lim, Y.P.; Kim, Y.K.; Hur, Y. Ogura-CMS in Chinese cabbage (Brassica rapa ssp. pekinensis) causes delayed expression of many nuclear genes. Plant Sci. 2013, 199, 7–12. [Google Scholar] [CrossRef]
- Du, K.; Liu, Q.; Wu, X.; Jiang, J.; Wu, J.; Fang, Y.; Li, A.; Wang, Y. Morphological Structure and Transcriptome Comparison of the Cytoplasmic Male Sterility Line in Brassica napus (SaNa-1A) Derived from Somatic Hybridization and Its Maintainer Line SaNa-1B. Front. Plant Sci. 2016, 7, 1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Kamiyama, M.; Iwata, T.; Makita, N.; Furukawa, H.; Ikeda, H. Moderate increase of mean daily temperature adversely affects fruit set of Lycopersicon esculentum by disrupting specific physiological processes in male reproductive development. Ann. Bot. 2006, 97, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Engelke, T.; Hirsche, J.; Roitsch, T. Anther-specific carbohydrate supply and restoration of metabolically engineered male sterility. J. Exp. Bot. 2010, 61, 2693–2706. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Liao, X.; Zhou, B.; Zhao, H.; Zhou, Y.; Zhou, R. Mutation in the coding sequence of atp6 are associated with male sterile cytoplasm in kenaf (Hibiscus cannabinus L.). Euphytica 2016, 207, 169–175. [Google Scholar] [CrossRef]
- Chen, C.; Chen, G.; Hao, X.; Cao, B.; Chen, Q.; Liu, S.; Lei, J. CaMF2, an anther-specific lipid transfer protein (LTP) gene, affects pollen development in Capsicum annuum L. Plant Sci. 2011, 181, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Wilson, Z.A.; Zhang, D.B. From arabidopsis to rice: Pathways in pollen development. J. Exp. Bot. 2009, 60, 1479–1492. [Google Scholar] [CrossRef] [Green Version]
- Phan, H.A.; Iacuone, S.; Li, S.F.; Parish, R.W. The MYB80 transcription factor is required for pollen development and the regulation of tapetal programmed cell death in Arabidopsis thaliana. Plant Cell 2011, 23, 2209–2224. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, A.M.; Kröber, S.; Unte, U.S.; Huijser, P.; Dekker, K.; Saedler, H. The Arabidopsis aborted microspores (ams) gene encodes a MYC class transcription factor. Plant J. 2003, 33, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Sun, Y.; Timofejeva, L.; Chen, C.; Grossniklaus, U.; Ma, H. Regulation of Arabidopsis tapetum development and function by Dysfunctional Tapetum1 (DYT1) encoding a putative bHLH transcription factor. Development 2006, 133, 3085–3095. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Yang, C.; Yuan, Z.; Zhang, D.; Gondwe, M.Y.; Ding, Z.; Liang, W.; Zhang, D.; Wilson, Z.A. The ABORTED MICROSPORES regulatory network is required for postmeiotic male reproductive development in Arabidopsis thaliana. Plant Cell 2010, 22, 91–107. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Yu, J.; Cheng, X.; Zong, X.; Xu, J.; Chen, M.; Li, Z.; Zhang, D.; Liang, W. The rice basic helix-loop-helix transcription factor TDR interacting protein2 is a central switch in early anther development. Plant Cell 2014, 26, 1512–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Tan, M.; Yu, H.; Li, L.; Zhou, F.; Yang, M.; Zhou, T.; Zhao, Y. Comparative transcriptome profiling of the fertile and sterile flower buds of a dominant genic male sterile line in sesame (Sesamum indicum L.). BMC Plant Biol. 2016, 16, 250. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.; Song, M.; Fan, S.; Yu, S. Transcriptomic analysis of differentially expressed genes during anther development in genetic male sterile and wild type cotton by digital gene-expression profiling. BMC Genom. 2013, 14, 97. [Google Scholar] [CrossRef] [Green Version]
- Ding, B.; Hao, M.; Mei, D.; Zaman, Q.; Sang, S.; Wang, H.; Wang, W.; Fu, L.; Cheng, H.; Hu, Q. Transcriptome and hormone comparison of three cytoplasmic male sterile systems in Brassica napus. Int. J. Mol. Sci. 2018, 19, 4022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, R.; Zhou, R.; Mmadi, M.A.; Li, D.; Qin, L.; Liu, A.; Wang, J.; Gao, Y.; Wei, M.; Shi, L.; et al. Root diversity in sesame (Sesamum indicum L.): Insights into the morphological, anatomical and gene expression profiles. Planta 2019, 250, 1461–1474. [Google Scholar] [CrossRef]
- Wang, L.; Dossa, K.; You, J.; Zhang, Y.; Li, D.; Zhou, R.; Yu, J.; Wei, X.; Zhu, X.; Jiang, S.; et al. High-resolution temporal transcriptome sequencing unravels ERF and WRKY as the master players in the regulatory networks underlying sesame responses to waterlogging and recovery. Genomics 2020, 113, 276–290. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. AgriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar]
- Ng, D.W.; Abeysinghe, J.K.; Kamali, M. Regulating the Regulators: The Control of Transcription Factors in Plant Defense Signaling. Int. J. Mol. Sci. 2018, 19, 3737. [Google Scholar] [CrossRef] [Green Version]
- Baillo, E.H.; Kimotho, R.N.; Zhang, Z.; Xu, P. Transcription Factors Associated with Abiotic and Biotic Stress Tolerance and Their Potential for Crops Improvement. Genes 2019, 10, 771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.K.; Zong, X.F.; Yu, G.D.; Li, J.N.; Zhang, W. Relationship between phytohormones and male sterility in thermo-photo-sensitive genic male sterile (TGMS) wheat. Euphytica 2006, 150, 241–248. [Google Scholar] [CrossRef]
- Sharma, K.D.; Nayyar, H. Regulatory networks in pollen development under cold stress. Front. Plant Sci. 2016, 7, 402. [Google Scholar] [CrossRef]
- Sundberg, E.; Østergaard, L. Distinct and dynamic auxin activities during reproductive development. Cold Spring Harb. Perspect. Biol. 2009, 1, a001628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Lv, Y.; Zhao, Y.; Nath, U.K.; Yuan, Y.; Wang, Z.; Yang, S.; Jia, H.; Wei, F.; Zhang, X. Comparative transcriptome analysis in Chinese cabbage (Brassica rapa ssp. pekinesis) for DEGs of Ogura-, Polima-CMS and their shared maintainer. Physiol. Mol. Biol. Plants. 2020, 26, 719–731. [Google Scholar] [CrossRef]
- Guedes, R.L.M.; Prosdocimi, F.; Fernandes, G.R.; Moura, L.K.; Ribeiro, H.A.L.; Ortega, J.M. Amino acids biosynthesis and nitrogen assimilation pathways: A great genomic deletion during eukaryotes evolution. BMC Genom. 2011, 12, S2. [Google Scholar] [CrossRef] [Green Version]
- Pei, X.; Jing, Z.; Tang, Z.; Zhu, Y. Comparative transcriptome analysis provides insight into differentially expressed genes related to cytoplasmic male sterility in broccoli (Brassica oleracea var. italica). Sci. Hortic. 2017, 217, 234–242. [Google Scholar] [CrossRef]
- Liu, J.; Pang, C.; Wei, H.; Song, M.; Meng, Y.; Ma, J.; Fan, S.; Yu, S. iTRAQ-facilitated proteomic profiling of anthers from a photosensitive male sterile mutant and wild-type cotton (Gossypium hirsutum L.). J. Proteom. 2015, 126, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhou, X.; Cao, Y.; Zhou, M.; McNeil, D.; Liang, S.; Yang, C. RNA-seq analysis of cold and drought responsive transcriptomes of Zea mays ssp. Mexicana L. Front. Plant Sci. 2017, 8, 136. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.W.; De Wang, C.; Cai, Q.Z.; Mei, S.Y.; Gao, L.; Zhou, Y.; Wang, T. Identification of promoter exchange at a male fertility restorer locus for cytoplasmic male sterility in radish (Raphanus sativus L.). Mol. Breed. 2017, 37, 82. [Google Scholar] [CrossRef]
- Brock, R.D.; Brock, R.D. Hormone-induced pear-apple hybrids. Heredity 1954, 8, 421–429. [Google Scholar] [CrossRef]
- Wu, J.; Qin, Y.; Zhao, J. Pollen tube growth is affected by exogenous hormones and correlated with hormone changes in styles in Torenia fournieri L. Plant Growth Regul. 2008, 55, 137–148. [Google Scholar] [CrossRef]
- Kwon, C.T.; Kim, S.H.; Kim, D.; Paek, N.C. The Rice Floral Repressor Early flowering1 Affects Spikelet Fertility By Modulating Gibberellin Signaling. Rice 2015, 8, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Cerny, R.E.; Qi, Y.; Bhat, D.; Aydt, C.M.; Hanson, D.D.; Malloy, K.P.; Ness, L.A. Transgenic studies on the involvement of cytokinin and gibberellin in male development. Plant Physiol. 2003, 131, 1270–1282. [Google Scholar] [CrossRef] [Green Version]
- Pike, L.M.; Peterson, C.E. Gibberellin A4/A7, for induction of staminate flowers on the gynoecious cucumber (Cucumis sativus L.). Euphytica 1969, 18, 106–109. [Google Scholar]
- Dai, S.; Kai, W.; Liang, B.; Wang, J.; Jiang, L.; Du, Y.; Sun, Y.; Leng, P. The functional analysis of SlNCED1 in tomato pollen development. Cell Mol. Life Sci. 2018, 75, 3457–3472. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Sawhney, V.K. Abscisic acid in a male sterile tomato mutant and its regulation by low temperature. J. Exp. Bot. 1998, 49, 199–203. [Google Scholar] [CrossRef]
- Mukhtar, M.S.; Liu, X.; Somssich, I.E. Elucidating the role of WRKY27 in male sterility in Arabidopsis. Plant Signal. Behav. 2017, 12, e1363945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Zhao, Z.; Liu, Y.; Liang, B.; Guan, S.; Lan, H.; Wang, J.; Lu, Y.; Cao, M. Comparative transcriptome analysis of isonuclear-alloplasmic lines unmask key transcription factor genes and metabolic pathways involved in sterility of maize CMS-C. Peer J. 2017, 5, e3408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, S.; Sahu, S.; Kaila, T.; Nigam, D.; Chaduvla, P.K.; Rao, A.R.; Sanand, S.; Singh, N.K.; Gaikwad, K. Transcriptome profiling of differentially expressed genes in cytoplasmic male-sterile line and its fertility restorer line in pigeon pea (Cajanus cajan L.). BMC Plant Biol. 2020, 20, 74. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Bao, S.; Zhou, X.; Liu, J.; Zhuang, Y. The key genes and pathways related to male sterility of eggplant revealed by comparative transcriptome analysis. BMC Plant Biol. 2018, 18, 209. [Google Scholar] [CrossRef]
- Goossens, J.; Mertens, J.; Goossens, A. Role and functioning of bHLH transcription factors in jasmonate signalling. J. Exp. Bot. 2017, 168, 1333–1347. [Google Scholar] [CrossRef]
- Fernández-Calvo, P.; Chini, A.; Fernández-Barbero, G.; Chico, J.M.; Gimenez-Ibanez, S.; Geerinck, J.; Eeckhout, D.; Schweizer, F.; Godoy, M.; Franco-Zorrilla, J.M.; et al. The Arabidopsis bHLH transcription factors MYC3 and MYC4 are targets of JAZ repressors and act additively with MYC2 in the activation of jasmonate responses. Plant Cell 2011, 23, 701–715. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Qi, T.; Huang, H.; Ren, Q.; Wu, D.; Chang, C.; Peng, W.; Liu, Y.; Peng, J.; Xie, D. The jasmonate-ZIM domain proteins interact with the R2R3-MYB transcription factors MYB21 and MYB24 to affect jasmonate-regulated stamen development in Arabidopsis. Plant Cell 2011, 23, 1000–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stracke, R.; Werber, M.; Weisshaar, B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 2001, 4, 447–456. [Google Scholar] [CrossRef]
- Preston, J.; Wheeler, J.; Heazlewood, J.; Li, S.F.; Parish, R.W. AtMYB32 is required for normal pollen development in Arabidopsis thaliana. Plant J. 2004, 40, 979–995. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, J.; Du, M.L.; Li, L.; Wang, X.L.; Li, X.B. A cotton gene encoding myb-like transcription factor is specifically expressed in pollen and is involved in regulation of late anther/pollen development. Plant Cell Physiol. 2013, 54, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Liu, T.; Wang, Z. Comparative transcriptome profile of the cytoplasmic male sterile and fertile floral buds of radish (Raphanus sativus L.). Int. J. Mol. Sci. 2016, 17, 42. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Ye, X.; Zhang, S.; Zhu, S.; Yuan, L.; Hou, J.; Wang, C. Comparative Transcriptome Analysis between Fertile and CMS Flower Buds in Wucai (Brassica campestris L.). BMC Genom. 2018, 19, 908. [Google Scholar] [CrossRef]
- Dossa, K.; Mmadi, M.A.; Zhou, R.; Zhang, T.; Su, R.; Zhang, Y.; Wang, L.; You, J.; Zhang, X. Depicting the Core Transcriptome Modulating Multiple Abiotic Stresses Responses in Sesame (Sesamum indicum L.). Int. J. Mol. Sci. 2019, 20, 3930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israelsen, W.J.; Vander Heiden, M.G. Pyruvate kinase: Function, regulation and role in cancer. Semin. Cell Dev. Biol. 2015, 43, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fucile, G.; Falconer, S.; Christendat, D. Evolutionary diversification of plant shikimate kinase gene duplicates. PLoS Genet. 2008, 4, e1000292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruprich-Robert, G.; Zickler, D.; Berteaux-Lecellier, V.; Vélot, C.; Picard, M. Lack of mitochondrial citrate synthase discloses a new meiotic checkpoint in a strict aerobe. EMBO J. 2002, 21, 6440–6451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Fu, H.F.; Gong, Z.H.; Chai, W.G. Involvement of a universal amino acid synthesis impediment in cytoplasmic male sterility in pepper. Sci. Rep. 2016, 6, 23357. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, J.; Lan, M.; Xu, X.; Yang, H.; Zhang, L.; Lv, F.; Yang, H.; Yang, D.; Li, C.; He, J. Transcriptome Profiling Reveals Molecular Changes during Flower Development between Male Sterile and Fertile Chinese Cabbage (Brassica rapa ssp. pekinensis) Lines. Life 2021, 11, 525. https://doi.org/10.3390/life11060525
Hu J, Lan M, Xu X, Yang H, Zhang L, Lv F, Yang H, Yang D, Li C, He J. Transcriptome Profiling Reveals Molecular Changes during Flower Development between Male Sterile and Fertile Chinese Cabbage (Brassica rapa ssp. pekinensis) Lines. Life. 2021; 11(6):525. https://doi.org/10.3390/life11060525
Chicago/Turabian StyleHu, Jingfeng, Mei Lan, Xuezhong Xu, Hongli Yang, Liqin Zhang, Fengxian Lv, Huiju Yang, Ding Yang, Chongjuan Li, and Jiangming He. 2021. "Transcriptome Profiling Reveals Molecular Changes during Flower Development between Male Sterile and Fertile Chinese Cabbage (Brassica rapa ssp. pekinensis) Lines" Life 11, no. 6: 525. https://doi.org/10.3390/life11060525
APA StyleHu, J., Lan, M., Xu, X., Yang, H., Zhang, L., Lv, F., Yang, H., Yang, D., Li, C., & He, J. (2021). Transcriptome Profiling Reveals Molecular Changes during Flower Development between Male Sterile and Fertile Chinese Cabbage (Brassica rapa ssp. pekinensis) Lines. Life, 11(6), 525. https://doi.org/10.3390/life11060525