
Proteomic Identification of Markers of Membrane Repair, Regeneration and Fibrosis in the Aged and Dystrophic Diaphragm

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Wild Type and Dystrophic Diaphragm Muscle Specimens
2.3. Preparation of Muscle Tissue Extracts
2.4. Mass Spectrometry and Proteomic Data Analysis
2.5. Comparative Immunoblot Analysis
3. Results
3.1. Mass Spectrometric Analysis of Young versus Aged mdx-4cv Diaphragm Muscle
3.2. Reduced Dystrophin Complex in mdx-4cv Diaphragm Muscle
3.3. Established Changes of Dystrophic Biomarkers in mdx-4cv Diaphragm Muscle
3.4. Proteomics of Excitation-Contraction Coupling and Calcium Handling in mdx-4cv Diaphragm
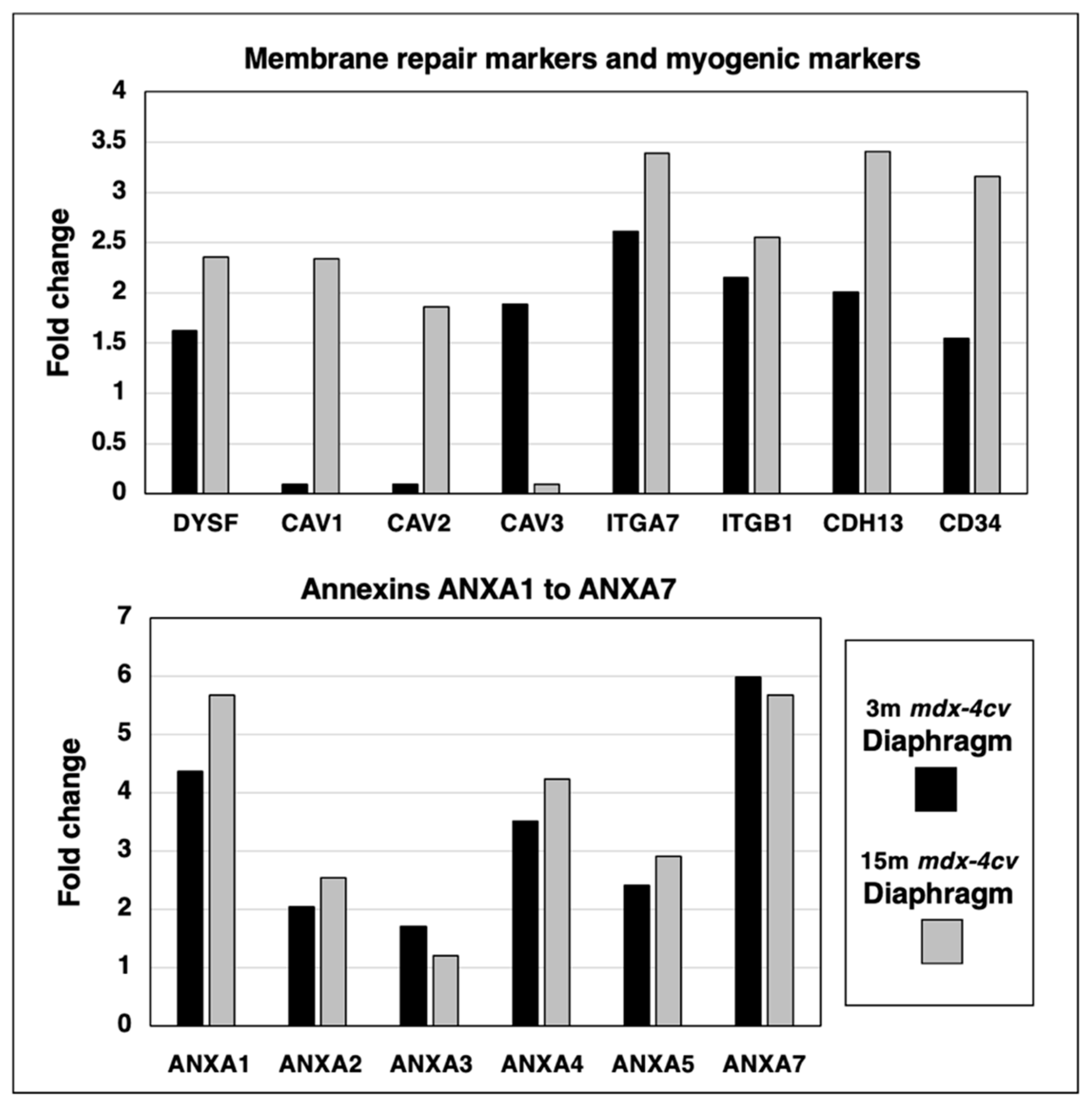
3.5. Proteomics of Membrane Repair and Calcium Sensing in mdx-4cv Diaphragm
3.6. Proteomics of Collagens and the Extracellular Matrix in mdx-4cv Diaphragm Muscle
3.7. Immunoblotting of Collagen VI, Periostin and Annexin 2 in mdx-4cv Diaphragm
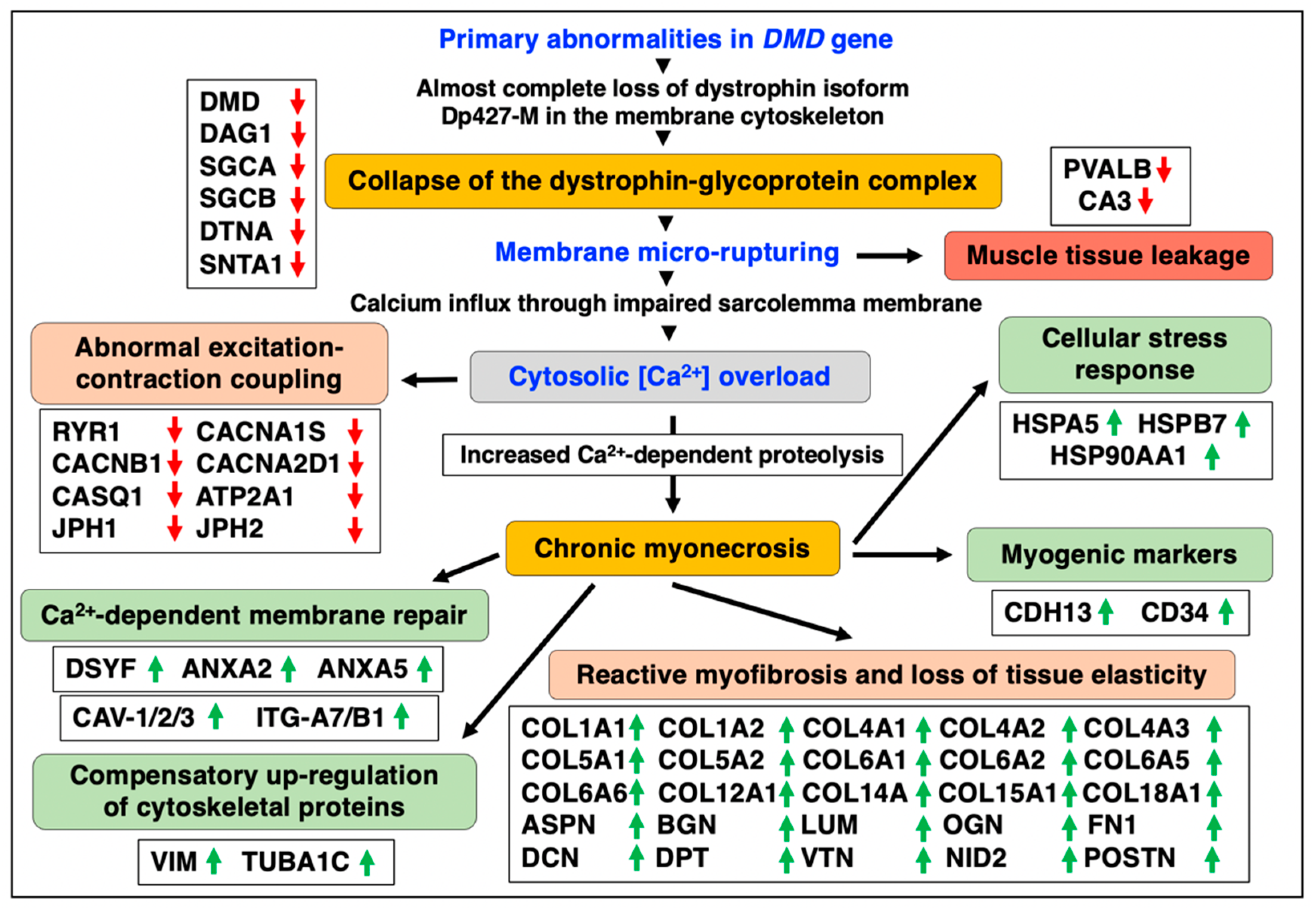
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Benarroch, L.; Bonne, G.; Rivier, F.; Hamroun, D. The 2020 version of the gene table of neuromuscular disorders. Neuromuscul. Disord. 2019, 29, 980–1018. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.; Spendiff, S.; Roos, A.; Bourque, P.R.; Warman Chardon, J.; Kirschner, J.; Horvath, V.; Lochmüller, H. Advances in the diagnosis of inherited neuromuscular diseases and implications for therapy development. Lancet Neurol. 2020, 19, 522–532. [Google Scholar] [CrossRef]
- Dowling, J.J.; Weihl, C.C.; Spencer, M.J. Molecular and cellular basis of genetically inherited skeletal muscle disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Prim. 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, E.; Samitt, C.E.; Miranda, A.F.; Hays, A.P.; Salviati, G.; DiMauro, S.; Kunkel, L.M.; Hoffman, E.P.; Rowland, L.P. Duchenne muscular dystrophy: Deficiency of dystrophin at the muscle cell surface. Cell 1988, 54, 447–452. [Google Scholar] [CrossRef]
- Hoffman, E.P. Causes of clinical variability in Duchenne and Becker muscular dystrophies and implications for exon skipping therapies. Acta Myol. 2020, 39, 179–186. [Google Scholar] [PubMed]
- Roberts, R.G. Dystrophin, its gene, and the dystrophinopathies. Adv. Genet. 1995, 33, 177–231. [Google Scholar] [PubMed]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Hudecki, M.S.; Rosenberg, P.A.; Pollina, C.M.; Kunkel, L.M. Cell and fiber-type distribution of dystrophin. Neuron 1988, 1, 411–420. [Google Scholar] [CrossRef]
- Delalande, O.; Czogalla, A.; Hubert, J.F.; Sikorski, A.; Le Rumeur, E. Dystrophin and Spectrin, Two Highly Dissimilar Sisters of the Same Family. Subcell. Biochem. 2017, 82, 373–403. [Google Scholar] [PubMed]
- Hoffman, E.P. The discovery of dystrophin, the protein product of the Duchenne muscular dystrophy gene. FEBS J. 2020, 287, 3879–3887. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K.; Campbell, K.P. Dystrophin constitutes 5% of membrane cytoskeleton in skeletal muscle. FEBS Lett. 1991, 283, 230–234. [Google Scholar] [CrossRef] [Green Version]
- Finn, D.M.; Ohlendieck, K. Rabbit brain and muscle isoforms containing the carboxy-terminal domain of 427 kDa skeletal muscle dystrophin exhibit similar biochemical properties. Neurosci. Lett. 1997, 222, 25–28. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Campbell, K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar] [CrossRef] [Green Version]
- Ervasti, J.M.; Campbell, K.P. Dystrophin and the membrane skeleton. Curr. Opin. Cell Biol. 1993, 5, 82–87. [Google Scholar] [CrossRef]
- Murphy, S.; Ohlendieck, K. The biochemical and mass spectrometric profiling of the dystrophin complexome from skeletal muscle. Comput. Struct. Biotechnol. J. 2015, 14, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Prins, K.W.; Humston, J.L.; Mehta, A.; Tate, V.; Ralston, E.; Ervasti, J.M. Dystrophin is a microtubule-associated protein. J. Cell Biol. 2009, 186, 363–369. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Murphy, S.; Zweyer, M.; Sabir, H.; Swandulla, D.; Ohlendieck, K. The Dystrophin Node as Integrator of Cytoskeletal Organization, Lateral Force Transmission, Fiber Stability and Cellular Signaling in Skeletal Muscle. Proteomes 2021, 9, 9. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Campbell, K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell 1991, 66, 1121–1131. [Google Scholar] [CrossRef]
- Ohlendieck, K. Towards an understanding of the dystrophin-glycoprotein complex: Linkage between the extracellular matrix and the membrane cytoskeleton in muscle fibers. Eur. J. Cell Biol. 1996, 69, 1–10. [Google Scholar] [PubMed]
- Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Gaver, M.G.; Campbell, K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990, 345, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K.; Campbell, K.P. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J. Cell Biol. 1991, 115, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- Ohlendieck, K.; Matsumura, K.; Ionasescu, V.V.; Towbin, J.A.; Bosch, E.P.; Weinstein, S.L.; Sernett, S.W.; Campbell, K.P. Duchenne muscular dystrophy: Deficiency of dystrophin-associated proteins in the sarcolemma. Neurology 1993, 43, 795–800. [Google Scholar] [CrossRef]
- Culligan, K.G.; Mackey, A.J.; Finn, D.M.; Maguire, P.B.; Ohlendieck, K. Role of dystrophin isoforms and associated proteins in muscular dystrophy (review). Int. J. Mol. Med. 1998, 2, 639–648. [Google Scholar] [CrossRef]
- Alderton, J.M.; Steinhardt, R.A. Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. J. Biol. Chem. 2000, 275, 9452–9460. [Google Scholar] [CrossRef] [Green Version]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [Green Version]
- Dowling, P.; Gargan, S.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of impaired excitation-contraction coupling and abnormal calcium handling in muscular dystrophy. Proteomics 2022, e2200003, in press. [Google Scholar] [CrossRef]
- Ohlendieck, K.; Swandulla, D. Complexity of skeletal muscle degeneration: Multi-systems pathophysiology and organ crosstalk in dystrophinopathy. Pflugers Arch. 2021, 473, 1813–1839. [Google Scholar] [CrossRef]
- Schultz, T.I.; Raucci, F.J., Jr.; Salloum, F.N. Cardiovascular Disease in Duchenne Muscular Dystrophy: Overview and Insight Into Novel Therapeutic Targets. JACC Basic Transl. Sci. 2022, 7, 608–625. [Google Scholar] [CrossRef]
- Tripodi, L.; Villa, C.; Molinaro, D.; Torrente, Y.; Farini, A. The Immune System in Duchenne Muscular Dystrophy Pathogenesis. Biomedicines 2021, 9, 1447. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic serum biomarkers for neuromuscular diseases. Expert Rev. Proteom. 2018, 15, 277–291. [Google Scholar] [CrossRef]
- Spitali, P.; Hettne, K.; Tsonaka, R.; Charrout, M.; van den Bergen, J.; Koeks, Z.; Kan, H.E.; Hooijmans, M.T.; Roos, A.; Straub, V.; et al. Tracking disease progression non-invasively in Duchenne and Becker muscular dystrophies. J. Cachexia Sarcopenia Muscle 2018, 9, 715–726. [Google Scholar] [CrossRef]
- Al-Khalili Szigyarto, C. Duchenne muscular dystrophy: Recent advances in protein biomarkers and the clinical application. Expert Rev. Proteom. 2020, 17, 365–375. [Google Scholar] [CrossRef]
- Carr, S.J.; Zahedi, R.P.; Lochmüller, H.; Roos, A. Mass spectrometry-based protein analysis to unravel the tissue pathophysiology in Duchenne muscular dystrophy. Proteom. Clin. Appl. 2018, 12, 1700071. [Google Scholar] [CrossRef]
- Dowling, P.; Murphy, S.; Zweyer, M.; Raucamp, M.; Swandulla, D.; Ohlendieck, K. Emerging proteomic biomarkers of X- linked muscular dystrophy. Expert Rev. Mol. Diagn. 2019, 19, 739–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zweyer, M.; Sabir, H.; Dowling, P.; Gargan, S.; Murphy, S.; Swandulla, D.; Ohlendieck, K. Histopathology of Duchenne muscular dystrophy in correlation with changes in proteomic biomarkers. Histol. Histopathol. 2022, 37, 101–116. [Google Scholar] [PubMed]
- Capitanio, D.; Moriggi, M.; Torretta, E.; Barbacini, P.; De Palma, S.; Viganò, A.; Capitanio, D.; Moriggi, M.; Torretta, E.; Barbacini, P.; et al. Comparative proteomic analyses of Duchenne muscular dystrophy and Becker muscular dystrophy muscles: Changes contributing to preserve muscle function in Becker muscular dystrophy patients. J. Cachexia Sarcopenia Muscle 2020, 11, 547–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capitanio, D.; Moriggi, M.; Barbacini, P.; Torretta, E.; Moroni, I.; Blasevich, F.; Morandi, L.; Mora, M.; Gelfi, C. Molecular fingerprint of BMD patients lacking a portion in the rod domain of dystrophin. Int. J. Mol. Sci. 2020, 23, 2624. [Google Scholar] [CrossRef] [PubMed]
- Doran, P.; Martin, G.; Dowling, P.; Jockusch, H.; Ohlendieck, K. Proteome analysis of the dystrophin-deficient MDX diaphragm reveals a drastic increase in the heat shock protein cvHSP. Proteomics 2006, 6, 4610–4621. [Google Scholar] [CrossRef]
- Guevel, L.; Lavoie, J.R.; Perez-Iratxeta, C.; Rouger, K.; Dubreil, L.; Feron, M.; Talon, S.; Brand, M.; Megeney, L.A. Quantitative proteomic analysis of dystrophic dog muscle. J. Proteome Res. 2011, 10, 2465–2478. [Google Scholar] [CrossRef]
- Rayavarapu, S.; Coley, W.; Cakir, E.; Jahnke, V.; Takeda, S.; Aoki, Y.; Grodish-Dressman, H.; Jaiswal, J.K.; Hoffman, E.P.; Brown, K.J.; et al. Identification of disease specific pathways using in vivo SILAC proteomics in dystrophin deficient mdx mouse. Mol. Cell Proteom. 2013, 12, 1061–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fröhlich, T.; Kemter, E.; Flenkenthaler, F.; Klymiuk, N.; Otte, K.A.; Blutke, A.; Krause, S.; Walter, M.C.; Wanke, R.; Wolf, E.; et al. Progressive muscle proteome changes in a clinically relevant pig model of Duchenne muscular dystrophy. Sci. Rep. 2016, 6, 33362. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Comparative gel-based proteomic analysis of chemically crosslinked complexes in dystrophic skeletal muscle. Electrophoresis 2018, 39, 1735–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Westering, T.L.E.; Johansson, H.J.; Hanson, B.; Coenen-Stass, A.M.L.; Lomonosova, Y.; Tanihata, J.; Motohashi, N.; Yokota, T.; Takeda, S.; Lehtiö, J.; et al. Mutation-independent proteomic signatures of pathological progression in murine models of Duchenne muscular dystrophy. Mol. Cell Proteom. 2020, 19, 2047–2068. [Google Scholar] [CrossRef] [PubMed]
- Partridge, T.A. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nghiem, P.P.; Kornegay, J.N. Gene therapies in canine models for Duchenne muscular dystrophy. Hum. Genet. 2019, 138, 483–489. [Google Scholar] [CrossRef]
- Zaynitdinova, M.I.; Lavrov, A.V.; Smirnikhina, S.A. Animal models for researching approaches to therapy of Duchenne muscular dystrophy. Transgenic Res. 2021, 30, 709–725. [Google Scholar] [CrossRef]
- Stirm, M.; Fonteyne, L.M.; Shashikadze, B.; Stöckl, J.B.; Kurome, M.; Keßler, B.; Zakhartchenko, V.; Kemter, E.; Blum, H.; Arnold, G.J.; et al. Pig models for Duchenne muscular dystrophy—From disease mechanisms to validation of new diagnostic and therapeutic concepts. Neuromuscul. Disord. 2022, 32, 543–556. [Google Scholar] [CrossRef]
- Markati, T.; Oskoui, M.; Farrar, M.A.; Duong, T.; Goemans, N.; Servais, L. Emerging therapies for Duchenne muscular dystrophy. Lancet Neurol. 2022, 21, 814–829. [Google Scholar] [CrossRef]
- Deng, J.; Zhang, J.; Shi, K.; Liu, Z. Drug development progress in duchenne muscular dystrophy. Front. Pharmacol. 2022, 13, 950651. [Google Scholar] [CrossRef] [PubMed]
- Eser, G.; Topaloğlu, H. Current Outline of Exon Skipping Trials in Duchenne Muscular Dystrophy. Genes 2022, 13, 1241. [Google Scholar] [CrossRef] [PubMed]
- Chung Liang, L.; Sulaiman, N.; Yazid, M.D. A Decade of Progress in Gene Targeted Therapeutic Strategies in Duchenne Muscular Dystrophy: A Systematic Review. Front. Bioeng. Biotechnol. 2022, 10, 833833. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Carberry, S.; Ohlendieck, K. Proteomics of the dystrophin-glycoprotein complex and dystrophinopathy. Curr. Protein Pept. Sci. 2013, 14, 680–697. [Google Scholar] [CrossRef] [PubMed]
- Brinkmeier, H.; Ohlendieck, K. Chaperoning heat shock proteins: Proteomic analysis and relevance for normal and dystrophin-deficient muscle. Proteom. Clin. Appl. 2014, 8, 875–895. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne muscular dystrophy: Myonecrosis, inflammation and oxidative stress. Dis. Model Mech. 2020, 13, dmm043638. [Google Scholar] [CrossRef] [Green Version]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef] [Green Version]
- Defour, A.; Medikayala, S.; Van der Meulen, J.H.; Hogarth, M.W.; Holdreith, N.; Malatras, A.; Duddy, W.; Boehler, J.; Nagaraju, K.; Jaiswal, J.K. Annexin A2 links poor myofiber repair with inflammation and adipogenic replacement of the injured muscle. Hum. Mol. Genet. 2017, 26, 1979–1991. [Google Scholar] [CrossRef] [Green Version]
- Bittel, D.C.; Chandra, G.; Tirunagri, L.M.S.; Deora, A.B.; Medikayala, S.; Scheffer, L.; Defour, A.; Jaiswal, J.K. Annexin A2 Mediates Dysferlin Accumulation and Muscle Cell Membrane Repair. Cells 2020, 9, 1919. [Google Scholar] [CrossRef]
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H., Jr. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003, 278, 50466–50473. [Google Scholar] [CrossRef] [Green Version]
- Han, R.; Campbell, K.P. Dysferlin and muscle membrane repair. Curr. Opin. Cell Biol. 2007, 19, 409–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demonbreun, A.R.; Rossi, A.E.; Alvarez, M.G.; Swanson, K.E.; Deveaux, H.K.; Earley, J.U.; Hadhazy, M.; Vohra, R.; Walter, G.A.; Pytel, P.; et al. Dysferlin and myoferlin regulate transverse tubule formation and glycerol sensitivity. Am. J. Pathol. 2014, 184, 248–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couchoux, H.; Bichraoui, H.; Chouabe, C.; Altafaj, X.; Bonvallet, R.; Allard, B.; Ronjat, M.; Berthier, C. Caveolin-3 is a direct molecular partner of the Cav1.1 subunit of the skeletal muscle L-type calcium channel. Int. J. Biochem. Cell Biol. 2011, 43, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, B.S.; Prószyński, T.J. A Role for Caveolin-3 in the Pathogenesis of Muscular Dystrophies. Int. J. Mol. Sci. 2020, 21, 8736. [Google Scholar] [CrossRef] [PubMed]
- Matsunobe, M.; Motohashi, N.; Aoki, E.; Tominari, T.; Inada, M.; Aoki, Y. Caveolin-3 regulates the activity of Ca2+/calmodulin-dependent protein kinase II in C2C12 cells. Am. J. Physiol. Cell Physiol. 2022, 323, C1137–C1148. [Google Scholar] [CrossRef]
- Collo, G.; Starr, L.; Quaranta, V. A new isoform of the laminin receptor integrin alpha 7 beta 1 is developmentally regulated in skeletal muscle. J. Biol. Chem. 1993, 268, 19019–19024. [Google Scholar] [CrossRef]
- Burkin, D.J.; Wallace, G.Q.; Milner, D.J.; Chaney, E.J.; Mulligan, J.A.; Kaufman, S.J. Transgenic expression of α7β1 integrin maintains muscle integrity, increases regenerative capacity, promotes hypertrophy, and reduces cardiomyopathy in dystrophic mice. Am. J. Pathol. 2005, 166, 253–263. [Google Scholar] [CrossRef]
- Liu, J.; Burkin, D.J.; Kaufman, S.J. Increasing alpha 7 beta 1-integrin promotes muscle cell proliferation, adhesion, and resistance to apoptosis without changing gene expression. Am. J. Physiol. Cell Physiol. 2008, 294, C627–C640. [Google Scholar] [CrossRef] [Green Version]
- Philippova, M.P.; Bochkov, V.N.; Stambolsky, D.V.; Tkachuk, V.A.; Resink, T.J. T-cadherin and signal-transducing molecules co-localize in caveolin-rich membrane domains of vascular smooth muscle cells. FEBS Lett. 1998, 429, 207–210. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kita, S.; Nishizawa, H.; Fukuda, S.; Fujishima, Y.; Obata, Y.; Nagao, H.; Masuda, S.; Nakamura, Y.; Shimizu, Y.; et al. Adiponectin promotes muscle regeneration through binding to T-cadherin. Sci. Rep. 2019, 9, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalbandian, M.; Zhao, M.; Sasaki-Honda, M.; Jonouchi, T.; Lucena-Cacace, A.; Mizusawa, T.; Yasuda, M.; Yoshida, Y.; Hotta, A.; Sakurai, H. Characterization of hiPSC-Derived Muscle Progenitors Reveals Distinctive Markers for Myogenic Cell Purification Toward Cell Therapy. Stem Cell Rep. 2021, 16, 883–898. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, J.R.; Heslop, L.; Yu, D.S.; Tajbakhsh, S.; Kelly, R.G.; Wernig, A.; Buckingham, M.E.; Partridge, T.A.; Zammit, P.S. Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. J. Cell Biol. 2000, 151, 1221–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankowski, R.J.; Deasy, B.M.; Cao, B.; Gates, C.; Huard, J. The role of CD34 expression and cellular fusion in the regeneration capacity of myogenic progenitor cells. J. Cell Sci. 2002, 115, 4361–4374. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Lázaro, D.; Garrosa, E.; Seco-Calvo, J.; Garrosa, M. Potential Satellite Cell-Linked Biomarkers in Aging Skeletal Muscle Tissue: Proteomics and Proteogenomics to Monitor Sarcopenia. Proteomes 2022, 10, 29. [Google Scholar] [CrossRef]
- Ito, N.; Miyagoe-Suzuki, Y.; Takeda, S.; Kudo, A. Periostin Is Required for the Maintenance of Muscle Fibers during Muscle Regeneration. Int. J. Mol. Sci. 2021, 22, 3627. [Google Scholar] [CrossRef]
- Ozdemir, C.; Akpulat, U.; Sharafi, P.; Yıldız, Y.; Onbaşılar, I.; Kocaefe, C. Periostin is temporally expressed as an extracellular matrix component in skeletal muscle regeneration and differentiation. Gene 2014, 553, 130–139. [Google Scholar] [CrossRef]
- Wang, Z.; An, J.; Zhu, D.; Chen, H.; Lin, A.; Kang, J.; Liu, W.; Kang, X. Periostin: An emerging activator of multiple signaling pathways. J. Cell Commun. Signal. 2022. [Google Scholar] [CrossRef]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [Green Version]
- Cescon, M.; Gattazzo, F.; Chen, P.; Bonaldo, P. Collagen VI at a glance. J. Cell Sci. 2015, 128, 3525–3531. [Google Scholar] [CrossRef]
- Cescon, M.; Gregorio, I.; Eiber, N.; Borgia, D.; Fusto, A.; Sabatelli, P.; Scorzeto, M.; Megighian, A.; Pegoraro, E.; Hashemolhosseini, S.; et al. Collagen VI is required for the structural and functional integrity of the neuromuscular junction. Acta Neuropathol. 2018, 136, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Dias, C.; Nylandsted, J. Plasma membrane integrity in health and disease: Significance and therapeutic potential. Cell Discov. 2021, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Croissant, C.; Carmeille, R.; Brévart, C.; Bouter, A. Annexins and Membrane Repair Dysfunctions in Muscular Dystrophies. Int. J. Mol. Sci. 2021, 22, 5276. [Google Scholar] [CrossRef] [PubMed]
- Croissant, C.; Gounou, C.; Bouvet, F.; Tan, S.; Bouter, A. Trafficking of Annexins during Membrane Repair in Human Skeletal Muscle Cells. Membranes 2022, 12, 153. [Google Scholar] [CrossRef]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Csapo, R.; Gumpenberger, M.; Wessner, B. Skeletal Muscle Extracellular Matrix—What Do We Know About Its Composition, Regulation, and Physiological Roles? A Narrative Review. Front. Physiol. 2020, 11, 253. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, Y.; Zhang, H. Extracellular matrix: An important regulator of cell functions and skeletal muscle development. Cell Biosci. 2021, 11, 65. [Google Scholar] [CrossRef]
- Desguerre, I.; Mayer, M.; Leturcq, F.; Barbet, J.P.; Gherardi, R.K.; Christov, C. Endomysial fibrosis in Duchenne muscular dystrophy: A marker of poor outcome associated with macrophage alternative activation. J. Neuropathol. Exp. Neurol. 2009, 68, 762–773. [Google Scholar] [CrossRef] [Green Version]
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 184–195. [Google Scholar]
- Ahmad, K.; Shaikh, S.; Ahmad, S.S.; Lee, E.J.; Choi, I. Cross-Talk Between Extracellular Matrix and Skeletal Muscle: Implications for Myopathies. Front. Pharmacol. 2020, 11, 142. [Google Scholar] [CrossRef] [Green Version]
- Holland, A.; Murphy, S.; Dowling, P.; Ohlendieck, K. Pathoproteomic profiling of the skeletal muscle matrisome in dystrophinopa- thy associated myofibrosis. Proteomics 2016, 16, 345–366. [Google Scholar] [CrossRef]
- Serrano, A.L.; Muñoz-Cánoves, P. Fibrosis development in early-onset muscular dystrophies: Mechanisms and translational implications. Semin. Cell Dev. Biol. 2017, 64, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K.; Swandulla, D. Molecular pathogenesis of Duchenne muscular dystrophy-related fibrosis. Pathologe 2017, 38, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Dowling, P.; Gargan, S.; Zweyer, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Protocol for the Bottom-Up Proteomic Analysis of Mouse Spleen. STAR Protoc. 2020, 1, 100196. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Concurrent Label-Free Mass Spectrometric Analysis of Dystrophin Isoform Dp427 and the Myofibrosis Marker Collagen in Crude Extracts from mdx-4cv Skeletal Muscles. Proteomes 2015, 3, 298–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.; Zweyer, M.; Raucamp, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of the mouse diaphragm and refined mass spectrometric analysis of the dystrophic phenotype. J. Muscle Res. Cell Motil. 2019, 40, 9–28. [Google Scholar] [CrossRef]
- Antharavally, B.S.; Mallia, K.A.; Rangaraj, P.; Haney, P.; Bell, P.A. Quantitation of proteins using a dye-metal-based colorimetric protein assay. Anal. Biochem. 2009, 385, 342–345. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [Green Version]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef] [PubMed]
- Duddy, W.; Duguez, S.; Johnston, H.; Cohen, T.V.; Phadke, A.; Gordish-Dressman, H.; Nagaraju, K.; Gnocchi, V.; Low, S.; Partridge, T. Muscular dystrophy in the mdx mouse is a severe myopathy compounded by hypotrophy, hypertrophy and hyperplasia. Skelet. Muscle 2015, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stedman, H.H.; Sweeney, H.L.; Shrager, J.B.; Maguire, H.C.; Panettieri, R.A.; Petrof, B.; Narusawa, M.; Leferovich, J.M.; Sladky, J.T.; Kelly, A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 1991, 352, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Sahani, R.; Wallace, C.H.; Jones, B.K.; Blemker, S.S. Diaphragm muscle fibrosis involves changes in collagen organization with mechanical implications in Duchenne muscular dystrophy. J. Appl. Physiol. 2022, 132, 653–672. [Google Scholar] [CrossRef] [PubMed]
- Giovarelli, M.; Arnaboldi, F.; Zecchini, S.; Cornaghi, L.B.; Nava, A.; Sommariva, M.; Clementi, E.G.I.; Gagliano, N. Characterisation of Progressive Skeletal Muscle Fibrosis in the Mdx Mouse Model of Duchenne Muscular Dystrophy: An In Vivo and In Vitro Study. Int. J. Mol. Sci. 2022, 23, 8735. [Google Scholar] [CrossRef]
- Im, W.B.; Phelps, S.F.; Copen, E.H.; Adams, E.G.; Slightom, J.L.; Chamberlain, J.S. Differential expression of dystrophin isoforms in strains of mdx mice with different mutations. Hum. Mol. Genet. 1996, 5, 1149–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, V.M.; Miller, D.R.; Armstrong, D.; Caskey, C.T. Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc. Natl. Acad. Sci. USA 1989, 86, 1292–1296. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.H.; Hakim, C.H.; Zhang, K.; Duan, D. Genotyping mdx, mdx3cv, and mdx4cv mice by primer competition polymerase chain reaction. Muscle Nerve 2011, 43, 283–286. [Google Scholar] [CrossRef] [Green Version]
- Danko, I.; Chapman, V.; Wolff, J.A. The frequency of revertants in mdx mouse genetic models for Duchenne muscular dystrophy. Pediatr. Res. 1992, 32, 128–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.; Henry, M.; Meleady, P.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Simultaneous Pathoproteomic Evaluation of the Dystrophin-Glycoprotein Complex and Secondary Changes in the mdx-4cv Mouse Model of Duchenne Muscular Dystrophy. Biology 2015, 4, 397–423. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Chemical crosslinking analysis of β-dystroglycan in dystrophin-deficient skeletal muscle. HRB Open Res. 2018, 1, 17. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic analysis of the sarcolemma-enriched fraction from dystrophic mdx-4cv skeletal muscle. J. Proteom. 2019, 191, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Dowling, P.; Zweyer, M.; Mundegar, R.R.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic analysis of dystrophin deficiency and associated changes in the aged mdx-4cv heart model of dystrophinopathy-related cardiomyopathy. J. Proteom. 2016, 145, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Gargan, S.; Dowling, P.; Zweyer, M.; Reimann, J.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Mass Spectrometric Profiling of Extraocular Muscle and Proteomic Adaptations in the mdx-4cv Model of Duchenne Muscular Dystrophy. Life 2021, 11, 595. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Dowling, P.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of mdx-4cv serum reveals highly elevated levels of the inflammation-induced plasma marker haptoglobin in muscular dystrophy. Int. J. Mol. Med. 2017, 39, 1357–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic identification of elevated saliva kallikrein levels in the mdx-4cv mouse model of Duchenne muscular dystrophy. Biochem. Biophys. Rep. 2018, 18, 100541. [Google Scholar] [CrossRef]
- Gargan, S.; Dowling, P.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Identification of marker proteins of muscular dystrophy in the urine proteome from the mdx-4cv model of dystrophinopathy. Mol. Omics 2020, 16, 268–278. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Label-free mass spectrometric analysis reveals complex changes in the brain proteome from the mdx-4cv mouse model of Duchenne muscular dystrophy. Clin. Proteom. 2015, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of liver tissue from the mdx-4cv mouse model of Duchenne muscular dystrophy. Clin. Proteom. 2018, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Gargan, S.; Zweyer, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteome-wide Changes in the mdx-4cv Spleen due to Pathophysiological Cross Talk with Dystrophin-Deficient Skeletal Muscle. iScience 2020, 23, 101500. [Google Scholar] [CrossRef]
- Dowling, P.; Zweyer, M.; Raucamp, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic and cell biological profiling of the renal phenotype of the mdx-4cv mouse model of Duchenne muscular dystrophy. Eur. J. Cell Biol. 2020, 99, 151059. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Gargan, S.; Zweyer, M.; Sabir, H.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of the interface between the stomach wall and the pancreas in dystrophinopathy. Eur. J. Transl. Myol. 2021, 31, 9627. [Google Scholar]
- Judge, L.M.; Haraguchiln, M.; Chamberlain, J.S. Dissecting the signaling and mechanical functions of the dystrophin-glycoprotein complex. J. Cell Sci. 2006, 119, 1537–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitrpant, C.; Fletcher, S.; Iversen, P.L.; Wilton, S.D. By-passing the nonsense mutation in the 4 CV mouse model of muscular dystrophy by induced exon skipping. J. Gene Med. 2009, 11, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Li, S.; Gregorevic, P.; Fall, B.M.; Chamberlain, J.S. Dystrophin delivery to muscles of mdx mice using lentiviral vectors leads to myogenic progenitor targeting and stable gene expression. Mol. Ther. 2010, 18, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Henry, M.; Meleady, P.; Winkler, C.K.; Krautwald, M.; Brinkmeier, H.; Ohlendieck, K. Comparative Label-Free Mass Spectrometric Analysis of Mildly versus Severely Affected mdx Mouse Skeletal Muscles Identifies Annexin, Lamin, and Vimentin as Universal Dystrophic Markers. Molecules 2015, 20, 11317–11344. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.; Brinkmeier, H.; Krautwald, M.; Henry, M.; Meleady, P.; Ohlendieck, K. Proteomic profiling of the dystrophin complex and membrane fraction from dystrophic mdx muscle reveals decreases in the cytolinker desmoglein and increases in the extracellular matrix stabilizers biglycan and fibronectin. J. Muscle Res. Cell Motil. 2017, 38, 251–268. [Google Scholar] [CrossRef] [Green Version]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Proteomics reveals drastic increase of extracellular matrix proteins collagen and dermatopontin in the aged mdx diaphragm model of Duchenne muscular dystrophy. Int. J. Mol. Med. 2012, 30, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Holland, A.; Dowling, P.; Meleady, P.; Henry, M.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Label-free mass spectrometric analysis of the mdx-4cv diaphragm identifies the matricellular protein periostin as a potential factor involved in dystrophinopathy-related fibrosis. Proteomics 2015, 15, 2318–2331. [Google Scholar] [CrossRef]
- Lorts, A.; Schwanekamp, J.A.; Baudino, T.A.; McNally, E.M.; Molkentin, J.D. Deletion of periostin reduces muscular dystrophy and fibrosis in mice by modulating the transforming growth factor-β pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 10978–10983. [Google Scholar] [CrossRef] [Green Version]
- Vontzalidis, A.; Terzis, G.; Manta, P. Increased dysferlin expression in Duchenne muscular dystrophy. Anal. Quant. Cytopathol. Histpathol. 2014, 36, 15–22. [Google Scholar] [PubMed]
- Boppart, M.D.; Mahmassani, Z.S. Integrin signaling: Linking mechanical stimulation to skeletal muscle hypertrophy. Am. J. Physiol. Cell Physiol. 2019, 317, C629–C641. [Google Scholar] [CrossRef] [PubMed]
- Burkin, D.J.; Wallace, G.Q.; Nicol, K.J.; Kaufman, D.J.; Kaufman, S.J. Enhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J. Cell Biol. 2001, 152, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Rooney, J.E.; Gurpur, P.B.; Burkin, D.J. Laminin-111 protein therapy prevents muscle disease in the mdx mouse model for Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 7991–7996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, K.N.; Montgomery, C.L.; Janssen, P.M.; Clark, K.R.; Mendell, J.R.; Rodino-Klapac, L.R. AAV-mediated overexpression of human α7 integrin leads to histological and functional improvement in dystrophic mice. Mol. Ther. 2013, 21, 520–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, J.L.; Oh, J.; Chou, E.; Lee, J.A.; Holmberg, J.; Burkin, D.J.; Crosbie-Watson, R.H. Sarcospan integration into laminin-binding adhesion complexes that ameliorate muscular dystrophy requires utrophin and α7 integrin. Hum. Mol. Genet. 2015, 24, 2011–2022. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Perlingeiro, R.C.R. Generation of human myogenic progenitors from pluripotent stem cells for in vivo regeneration. Cell. Mol. Life Sci. 2022, 79, 406. [Google Scholar] [CrossRef]
- Szigyarto, C.A.; Spitali, P. Biomarkers of Duchenne muscular dystrophy: Current findings. Degener. Neurol. Neuromuscul. Dis. 2018, 8, 1–13. [Google Scholar]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gargan, S.; Dowling, P.; Zweyer, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic Identification of Markers of Membrane Repair, Regeneration and Fibrosis in the Aged and Dystrophic Diaphragm. Life 2022, 12, 1679. https://doi.org/10.3390/life12111679
Gargan S, Dowling P, Zweyer M, Henry M, Meleady P, Swandulla D, Ohlendieck K. Proteomic Identification of Markers of Membrane Repair, Regeneration and Fibrosis in the Aged and Dystrophic Diaphragm. Life. 2022; 12(11):1679. https://doi.org/10.3390/life12111679
Chicago/Turabian StyleGargan, Stephen, Paul Dowling, Margit Zweyer, Michael Henry, Paula Meleady, Dieter Swandulla, and Kay Ohlendieck. 2022. "Proteomic Identification of Markers of Membrane Repair, Regeneration and Fibrosis in the Aged and Dystrophic Diaphragm" Life 12, no. 11: 1679. https://doi.org/10.3390/life12111679
APA StyleGargan, S., Dowling, P., Zweyer, M., Henry, M., Meleady, P., Swandulla, D., & Ohlendieck, K. (2022). Proteomic Identification of Markers of Membrane Repair, Regeneration and Fibrosis in the Aged and Dystrophic Diaphragm. Life, 12(11), 1679. https://doi.org/10.3390/life12111679