


An In-Silico Identification of Potential Flavonoids against Kidney Fibrosis Targeting TGFβR-1

 ,
,  ,
,  ,
,  , , ,
, , ,  , and
, and 
Abstract
:1. Introduction
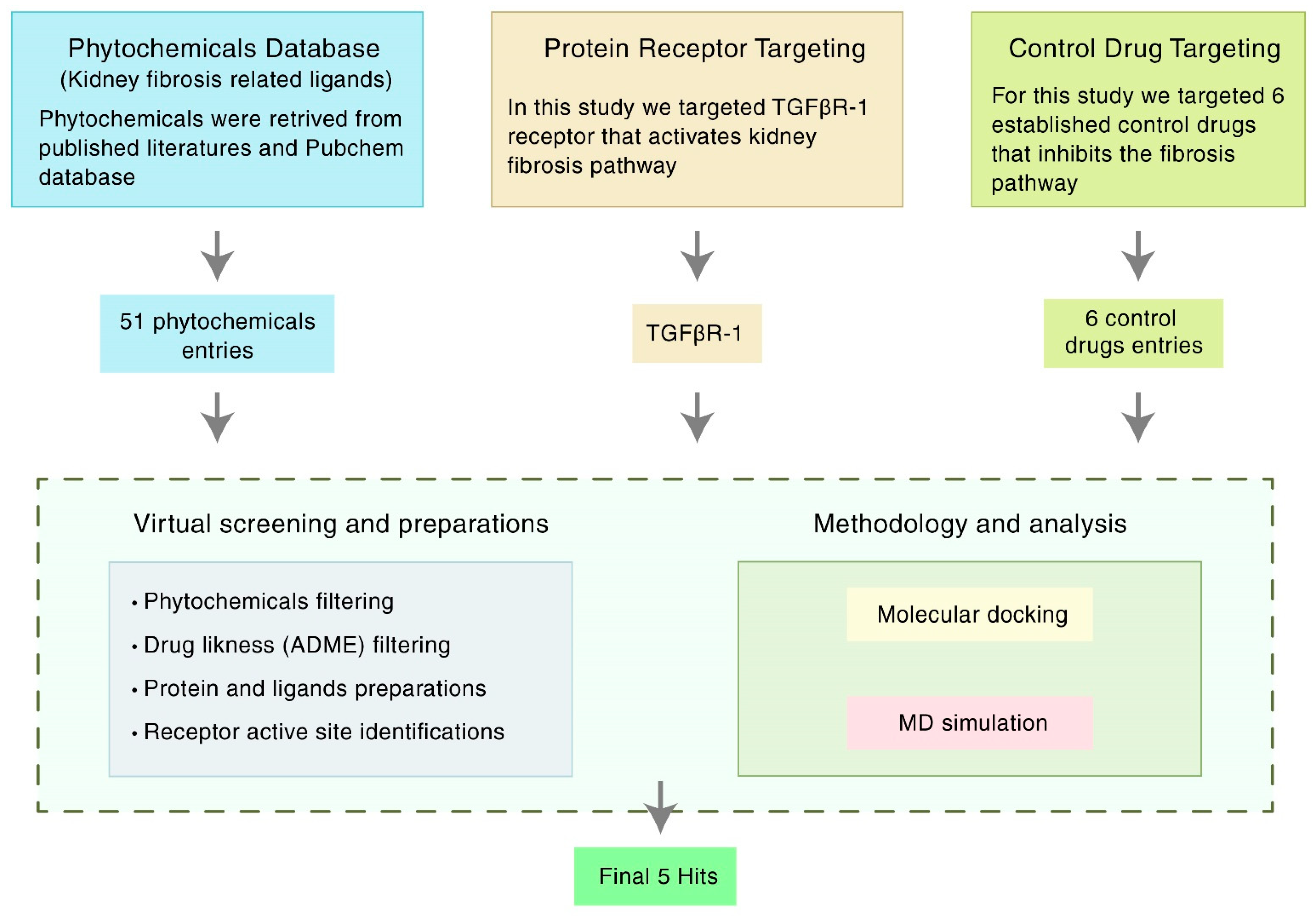
2. Materials and Methods
2.1. Receptor and Ligand Selection
2.2. Compound Screening and ADMET Prediction
2.3. Protein Preparation
2.4. Ligand Preparation
2.5. Active Site (ASs) Identification and Receptor Grid Generation
2.6. Molecular Docking and Visualization
2.7. Binding Free Energy Calculation by Using Prime/MM-GBSA Approach
2.8. Molecular Dynamics (MD) Simulations
2.8.1. Simulation Trajectory Analysis
2.8.2. RMSD Analysis
3. Results
3.1. Compound Screening and ADME/T
3.2. AS Identification and Receptor Grid Generation
3.3. Molecular Docking and MMGBSA Analysis
3.4. Analysis of Protein–Ligand Interactions
3.5. MD Simulations Analysis
3.5.1. RMSD Analysis
3.5.2. Protein–Ligand Contacts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castro, A.F.; Coresh, J. CKD surveillance using laboratory data from the population-based National Health and Nutrition Examination Survey (NHANES). Am. J Kidney Dis. 2009, 53, S46–S55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djudjaj, S.; Boor, P. Cellular and molecular mechanisms of kidney fibrosis. Mol. Asp. Med. 2019, 65, 16–36. [Google Scholar] [CrossRef] [PubMed]
- López-Novoa, J.M.; Martínez-Salgado, C.; Rodríguez-Peña, A.B.; López-Hernández, F.J. Common pathophysiological mechanisms of chronic kidney disease: Therapeutic perspectives. Pharmacol. Ther. 2010, 128, 61–81. [Google Scholar] [CrossRef]
- Izuhara, Y.; Nangaku, M.; Inagi, R.; Tominaga, N.; Aizawa, T.; Kurokawa, K.; van Ypersele de Strihou, C.; Miyata, T. Renoprotective properties of angiotensin receptor blockers beyond blood pressure lowering. J. Am. Soc. Nephrol. 2005, 16, 3631–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. TGF-β/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Kim, S.I.; Choi, M.E. Therapeutic targets for treating fibrotic kidney diseases. Transl. Res. J. Lab. Clin. Med. 2015, 165, 512–530. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J. Am. Soc. Nephrol. 2010, 21, 212–222. [Google Scholar] [CrossRef] [Green Version]
- Ungefroren, H.; Gieseler, F.; Kaufmann, R.; Settmacher, U.; Lehnert, H.; Rauch, B.H. Signaling Crosstalk of TGF-β/ALK5 and PAR2/PAR1: A Complex Regulatory Network Controlling Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1568. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.; Liang, P.; Liu, B.; Xu, Z.; Zhang, L.; Feng, M.; Tang, Y.; Xu, A. MicroRNA-140-5p Mediates Renal Fibrosis Through TGF-β1/Smad Signaling Pathway by Directly Targeting TGFBR1. Front. Physiol. 2020, 11, 1093. [Google Scholar] [CrossRef]
- Qi, W.; Chen, X.; Poronnik, P.; Pollock, C.A. Transforming growth factor-beta/connective tissue growth factor axis in the kidney. Int. J. Biochem. Cell Biol. 2008, 40, 9–13. [Google Scholar] [CrossRef]
- Wang, W.; Koka, V.; Lan, H.Y. Transforming growth factor-beta and Smad signalling in kidney diseases. Nephrology (Carlton) 2005, 10, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Hemshekhar, M.; Sunitha, K.; Santhosh, M.S.; Devaraja, S.; Kemparaju, K.; Vishwanath, B.; Niranjana, S.; Girish, K.J.P.R. An overview on genus Garcinia: Phytochemical and therapeutical aspects. Phytochem. Rev. 2011, 10, 325–351. [Google Scholar] [CrossRef]
- Shuaib, M.; Kushwaha, P.P.; Prajapati, K.S.; Singh, A.K.; Sharma, R.; Kumar, S. Effect of Dietary Phytochemicals in Obesity and Cancer. In Obesity and Cancer; Springer: Berlin/Heidelberg, Germany, 2021; pp. 163–184. [Google Scholar]
- Pagliaro, B.; Santolamazza, C.; Simonelli, F.; Rubattu, S. Phytochemical Compounds and Protection from Cardiovascular Diseases: A State of the Art. BioMed Res. Int. 2015, 2015, 918069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castañeda, R.; Cáceres, A.; Cruz, S.M.; Aceituno, J.A.; Marroquín, E.S.; Barrios Sosa, A.C.; Strangman, W.K.; Williamson, R.T. Nephroprotective plant species used in traditional Mayan Medicine for renal-associated diseases. J. Ethnopharmacol. 2022, 301, 115755. [Google Scholar] [CrossRef]
- Abd-Elkareem, M.; Soliman, M.; Abd El-Rahman, M.A.M.; Abou Khalil, N.S. Effect of Nigella sativa L. Seed on the Kidney of Monosodium Glutamate Challenged Rats. Front. Pharmacol. 2022, 13, 789988. [Google Scholar] [CrossRef]
- Rahman, M.A.; Akter, S.; Dorotea, D.; Mazumder, A.; Uddin, M.N.; Hannan, M.A.; Hossen, M.J.; Ahmed, M.S.; Kim, W.; Kim, B.; et al. Renoprotective potentials of small molecule natural products targeting mitochondrial dysfunction. Front. Pharmacol. 2022, 13, 925993. [Google Scholar] [CrossRef]
- Akter, S.; Moni, A.; Faisal, G.M.; Uddin, M.R.; Jahan, N.; Hannan, M.A.; Rahman, A.; Uddin, M.J. Renoprotective Effects of Mangiferin: Pharmacological Advances and Future Perspectives. Int. J. Environ. Res. Public Health 2022, 19, 1864. [Google Scholar] [CrossRef]
- Akter, T.; Rahman, M.A.; Moni, A.; Apu, M.A.I.; Fariha, A.; Hannan, M.A.; Uddin, M.J. Prospects for Protective Potential of Moringa oleifera against Kidney Diseases. Plants (Basel) 2021, 10, 2818. [Google Scholar] [CrossRef]
- Vargas, F.; Romecín, P.; García-Guillén, A.I.; Wangesteen, R.; Vargas-Tendero, P.; Paredes, M.D.; Atucha, N.M.; García-Estañ, J. Flavonoids in Kidney Health and Disease. Front. Physiol. 2018, 9, 394. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Ding, X.; Ding, Z.; Jia, P. Total Flavonoids from Leaves of Carya Cathayensis Ameliorate Renal Fibrosis via the miR-21/Smad7 Signaling Pathway. Cell. Physiol. Biochem. 2018, 49, 1551–1563. [Google Scholar] [CrossRef]
- Chen, H.A.; Chen, C.M.; Guan, S.S.; Chiang, C.K.; Wu, C.T.; Liu, S.H. The antifibrotic and anti-inflammatory effects of icariin on the kidney in a unilateral ureteral obstruction mouse model. Phytomedicine 2019, 59, 152917. [Google Scholar] [CrossRef] [PubMed]
- Ganbold, M.; Shimamoto, Y.; Ferdousi, F.; Tominaga, K.; Isoda, H. Antifibrotic effect of methylated quercetin derivatives on TGFβ-induced hepatic stellate cells. Biochem. Biophys. Rep. 2019, 20, 100678. [Google Scholar] [CrossRef]
- Pan, X.; Ma, X.; Jiang, Y.; Wen, J.; Yang, L.; Chen, D.; Cao, X.; Peng, C. A Comprehensive Review of Natural Products against Liver Fibrosis: Flavonoids, Quinones, Lignans, Phenols, and Acids. Evid. Based Complementary Altern. Med. 2020, 2020, 7171498. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.G.; Lu, X.H.; Li, W.; Zhao, X.; Zhang, C. Protective Effects of Luteolin on Diabetic Nephropathy in STZ-Induced Diabetic Rats. Evid.-Based Complementary Altern. Med. Ecam 2011, 2011, 323171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Hartogh, D.J.; Tsiani, E. Health Benefits of Resveratrol in Kidney Disease: Evidence from In Vitro and In Vivo Studies. Nutrients 2019, 11, 1624. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lu, H.; Xie, S.; Wu, C.; Guo, Y.; Xiao, Y.; Zheng, S.; Zhu, H.; Zhang, Y.; Bai, Y. Resveratrol suppresses the myofibroblastic phenotype and fibrosis formation in kidneys via proliferation-related signalling pathways. Br. J. Pharmacol. 2019, 176, 4745–4759. [Google Scholar] [CrossRef]
- Prince, P.D.; Fraga, C.G.; Galleano, M. (-)-Epicatechin administration protects kidneys against modifications induced by short-term l-NAME treatment in rats. Food Funct. 2020, 11, 318–327. [Google Scholar] [CrossRef]
- Yao, L.; Li, L.; Li, X.; Li, H.; Zhang, Y.; Zhang, R.; Wang, J.; Mao, X. The anti-inflammatory and antifibrotic effects of Coreopsis tinctoria Nutt on high-glucose-fat diet and streptozotocin-induced diabetic renal damage in rats. BMC Complementary Altern. Med. 2015, 15, 314. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Li, J.; Li, L.; Li, X.; Zhang, R.; Zhang, Y.; Mao, X. Coreopsis tinctoria Nutt ameliorates high glucose-induced renal fibrosis and inflammation via the TGF-β1/SMADS/AMPK/NF-κB pathways. BMC Complementary Altern. Med. 2019, 19, 14. [Google Scholar] [CrossRef]
- Lai, P.-B.; Zhang, L.; Yang, L.-Y. Quercetin Ameliorates Diabetic Nephropathy by Reducing the Expressions of Transforming Growth Factor-β1 and Connective Tissue Growth Factor in Streptozotocin-Induced Diabetic Rats. Ren. Fail. 2012, 34, 83–87. [Google Scholar] [CrossRef]
- Harikrishnan, L.S.; Warrier, J.; Tebben, A.J.; Tonukunuru, G.; Madduri, S.R.; Baligar, V.; Mannoori, R.; Seshadri, B.; Rahaman, H.; Arunachalam, P.N.; et al. Heterobicyclic inhibitors of transforming growth factor beta receptor I (TGFβRI). Bioorganic Med. Chem. 2018, 26, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Qu, J.; Wang, J.; Han, K.; Li, Q.; Bi, W.; Liu, R. Discovery of pulmonary fibrosis inhibitor targeting TGF-β RI in Polygonum cuspidatum by high resolution mass spectrometry with in silico strategy. J. Pharm. Anal. 2020, in press. [CrossRef]
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Modeling 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T. New Method for Fast and Accurate Binding-site Identification and Analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef]
- Goodford, P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef]
- Weber, A.E.; Halgren Ta Fau-Doyle, J.J.; Doyle Jj Fau-Lynch, R.J.; Lynch Rj Fau-Siegl, P.K.; Siegl Pk Fau-Parsons, W.H.; Parsons Wh Fau-Greenlee, W.J.; Greenlee Wj Fau-Patchett, A.A.; Patchett, A.A. Design and synthesis of P2-P1′-linked macrocyclic human renin inhibitors. J. Med. Chem. 1991, 34, 2692–2701. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Thérien, M.; Skorey, K.; Fau-Zamboni, R.; Zamboni, R.; Fau-Li, C.S.; Li Cs Fau-Lau, C.K.; Lau Ck Fau-LeRiche, T.; LeRiche T Fau-Linh Truong, V.; Linh Truong, V.; Fau-Waddleton, D.; et al. Synthesis of a novel peptidic photoaffinity probe for the PTP-1B enzyme. Bioorganic Med. Chem. Lett. 2004, 14, 2319–2322. [Google Scholar] [CrossRef]
- Hossain, R.; Al-Khafaji, K.; Khan, R.A.; Sarkar, C.; Islam, M.; Dey, D.; Jain, D.; Faria, F.; Akbor, R.; Atolani, O.J.P. Quercetin and/or Ascorbic Acid Modulatory Effect on Phenobarbital-Induced Sleeping Mice Possibly through GABAA and GABAB Receptor Interaction Pathway. Pharmaceuticals (Basel) 2021, 14, 721. [Google Scholar] [CrossRef]
- Hoffman, J.M.; Margolis, K.G. Building community in the gut: A role for mucosal serotonin. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 6–8. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Dubey, A.; Yadava, U.; Mishra, S.K.; Kang, S.G.; Dwivedi, V.D. Exploration of natural compounds with anti-SARS-CoV-2 activity via inhibition of SARS-CoV-2 Mpro. Brief. Bioinform. 2021, 22, 1361–1377. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wang, Z.; Ren, J.; Wei, Z.; Li, J. Potential inhibitors for the novel coronavirus (SARS-CoV-2). Brief. Bioinform. 2021, 22, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Shawky, E. Prediction of potential cancer-related molecular targets of North African plants constituents using network pharmacology-based analysis. J. Ethnopharmacol. 2019, 238, 111826. [Google Scholar] [CrossRef] [PubMed]
- Ahammad, F.; Alam, R.; Mahmud, R.; Akhter, S.; Talukder, E.K.; Tonmoy, A.M.; Fahim, S.; Al-Ghamdi, K.; Samad, A.; Qadri, I. Pharmacoinformatics and molecular dynamics simulation-based phytochemical screening of neem plant (Azadiractha indica) against human cancer by targeting MCM7 protein. Brief. Bioinform. 2021, 22, bbab098. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Deng, S.; Koneski, I.; Awad, M.M.; Akram, Z.; Matinlinna, J.; Pichika, M.R.; Daood, U.; Fawzy, A.S. Multiscale in-vitro analysis of photo-activated riboflavin incorporated in an experimental universal adhesive. J. Mech. Behav. Biomed. Mater. 2020, 112, 104082. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Sarkar, B.; Ullah, M.A.; Islam, S.S.; Rahman, M.D.H.; Araf, Y. Analysis of plant-derived phytochemicals as anti-cancer agents targeting cyclin dependent kinase-2, human topoisomerase IIa and vascular endothelial growth factor receptor-2. J. Recept. Signal Transduct. 2021, 41, 217–233. [Google Scholar] [CrossRef]
- Lenselink, E.B.; Louvel, J.; Forti, A.F.; van Veldhoven, J.P.D.; de Vries, H.; Mulder-Krieger, T.; McRobb, F.M.; Negri, A.; Goose, J.; Abel, R.; et al. Predicting Binding Affinities for GPCR Ligands Using Free-Energy Perturbation. ACS Omega 2016, 1, 293–304. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.-m.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Dey, D.; Paul, P.K.; Al Azad, S.; Al Mazid, M.F.; Khan, A.M.; Sharif, M.A.; Rahman, M.H. Molecular optimization, docking, and dynamic simulation profiling of selective aromatic phytochemical ligands in blocking the SARS-CoV-2 S protein attachment to ACE2 receptor: An in silico approach of targeted drug designing. J. Adv. Vet. Anim. Res. 2021, 8, 24–35. [Google Scholar] [CrossRef]
- Jain, D.; Hossain, R.; Khan, R.A.; Dey, D.; Toma, T.R.; Islam, M.T.; Janmeda, P.; Hakeem, K.R. Computer-aided Evaluation of Anti-SARS-CoV-2 (3-chymotrypsin-like Protease and Transmembrane Protease Serine 2 Inhibitors) Activity of Cepharanthine: An In silico Approach. Biointerface Res. Appl. Chem. 2022, 12, 768–780. [Google Scholar]
- Mannhold, R.; Kubinyi, H.; Folkers, G. Pharmacokinetics and Metabolism in Drug Design, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2012; Volume 51. [Google Scholar]
- Notari, R.E. Pharmacokinetics Molecular Modification: Implications in Drug Design Evaluation. J. Pharm. Sci. 1973, 62, 865–881. [Google Scholar] [CrossRef] [PubMed]
- Sharif, M.A.; Hossen, M.S.; Shaikat, M.M.; Mashuk, F.; Haidary, T.I.E.; Dey, D.; Paul, P.K.; Al Azad, S.; Al Mazid, M.F.; Badal, M.N.U. Molecular Optimization, Docking and Dynamic Simulation Study of Selective Natural Aromatic Components to Block E2-CD81 Complex Formation in Predating Protease Inhibitor Resistant HCV Influx. Int. J. Pharm. Res. 2021, 13, 3512–3525. [Google Scholar]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins Struct. Funct. Bioinform. 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, K.; Grauffel, C.D.; Lim, C. How molecular size impacts RMSD applications in molecular dynamics simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | PubChem CID | Chemical Name | Chemical Structure | Docking Scores (kcal/mol) | MMGBSA Scores (kcal/mol) |
|---|---|---|---|---|---|
| C-06 | 72,276 | (-)-Epicatechin |  | −13.585 | −48.19 |
| C-18 | 5,281,614 | Fisetin |  | −13.177 | −51.84 |
| C-29 | 5,280,445 | Luteolin |  | −10.506 | −44.80 |
| C-09 | 445,154 | Resveratrol |  | −8.552 | −43.34 |
| C-01 | 136,417 | 5-Hydroxy-3,6,7,8,3′,4′-hexamethoxyflavone |  | −7.808 | −47.97 |
| C-32 | 10,096,344 | Linagliptin (control) |  | −9.074 | −41.57 |
| PubChem ID | Residues | Distance(Å) | Bond Category | Bond Type |
|---|---|---|---|---|
| CID-72276 | Asp351 | 4.15 | Hydrogen | Conventional Hydrogen |
| Tyr249 | 5.64 | Hydrogen | Conventional Hydrogen | |
| Glu245 | 4.82 | Hydrogen | Conventional Hydrogen | |
| His283 | 4.08 | Hydrogen | Conventional Hydrogen | |
| Ala350 | 5.36 | Hydrophobic | Pi–Alkyl | |
| Leu260 | 4.98 | Hydrophobic | Pi–Alkyl | |
| Ile211 | 4.63 | Hydrophobic | Pi–Alkyl | |
| Ala230 | 4.57 | Hydrophobic | Pi–Alkyl | |
| Val219 | 5.08 | Hydrophobic | Pi–Alkyl | |
| Leu340 | 4.58 | Hydrophobic | Pi–Alkyl | |
| Lys232 | 4.17 | Hydrophobic | Pi–Cation | |
| CID-5281614 | Glu245 | 2.7 | Hydrogen | Conventional Hydrogen |
| Ala350 | 2.81 | Hydrogen | Carbon–Hydrogen | |
| Ala230 | 4.13 | Hydrophobic | Pi–Alkyl | |
| Leu340 | 4.71 | Hydrophobic | Pi–Alkyl | |
| Leu260 | 4.92 | Hydrophobic | Pi–Alkyl | |
| Ile211 | 2.76 | Hydrophobic | Pi–Sigma | |
| Val219 | 2.68 | Hydrophobic | Pi–Sigma | |
| Lys232 | 3.85 | Hydrophobic | Pi–Cation | |
| CID-5280445 | Gly286 | 3.61 | Hydrogen | Carbon–Hydrogen |
| Val219 | 4.34 | Hydrophobic | Pi–Alkyl | |
| Ala350 | 4.74 | Hydrophobic | Pi–Alkyl | |
| Ala230 | 5.24 | Hydrophobic | Pi–Alkyl | |
| Lys232 | 4.41 | Hydrophobic | Pi–Cation | |
| CID-445154 | Ile211 | 4.78 | Hydrophobic | Pi–Alkyl |
| CID-136417 | Lys232 | 2.37 | Hydrogen | Conventional Hydrogen |
| Ser280 | 2.37 | Hydrogen | Conventional Hydrogen | |
| Ala350 | 2.95 | Hydrogen | Carbon–Hydrogen | |
| Ala230 | 4.57 | Hydrophobic | Pi–Alkyl |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.H.; Biswas, P.; Dey, D.; Hannan, M.A.; Sahabuddin, M.; Araf, Y.; Kwon, Y.; Emran, T.B.; Ali, M.S.; Uddin, M.J. An In-Silico Identification of Potential Flavonoids against Kidney Fibrosis Targeting TGFβR-1. Life 2022, 12, 1764. https://doi.org/10.3390/life12111764
Rahman MH, Biswas P, Dey D, Hannan MA, Sahabuddin M, Araf Y, Kwon Y, Emran TB, Ali MS, Uddin MJ. An In-Silico Identification of Potential Flavonoids against Kidney Fibrosis Targeting TGFβR-1. Life. 2022; 12(11):1764. https://doi.org/10.3390/life12111764
Chicago/Turabian StyleRahman, MD. Hasanur, Partha Biswas, Dipta Dey, Md. Abdul Hannan, Md. Sahabuddin, Yusha Araf, Youngjoo Kwon, Talha Bin Emran, Md. Sarafat Ali, and Md Jamal Uddin. 2022. "An In-Silico Identification of Potential Flavonoids against Kidney Fibrosis Targeting TGFβR-1" Life 12, no. 11: 1764. https://doi.org/10.3390/life12111764
APA StyleRahman, M. H., Biswas, P., Dey, D., Hannan, M. A., Sahabuddin, M., Araf, Y., Kwon, Y., Emran, T. B., Ali, M. S., & Uddin, M. J. (2022). An In-Silico Identification of Potential Flavonoids against Kidney Fibrosis Targeting TGFβR-1. Life, 12(11), 1764. https://doi.org/10.3390/life12111764