


Prophylactic Treatment with Hydrogen Sulphide Can Prevent Renal Ischemia-Reperfusion Injury in L-NAME Induced Hypertensive Rats with Cisplatin-Induced Acute Renal Failure

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Drugs
2.2. Ethical Statement and Experimental Groups
2.3. Induction of ARF, Hypertension, Adminiistration of NaHS and Metabolic Data Collection
2.4. Measurement of NIBP
2.5. Acute Surgical Experiment
2.5.1. Collection of Urine and Blood Samples
2.5.2. Induction of Ischemia-Reperfusion
2.6. Measurement of Plasma H2S Concentration
2.7. Renal Function Parameters Measurement
2.8. Oxidative Stress Parameters Measurement
2.9. Measurement of Plasma and Kidney Tissue Concentrations of ICAM-1 and NF-kB
2.10. Histopathology of Kidney by Using Hematoxylin and Eosin (H&E) Staining
2.11. Statistical Analysis
3. Results
3.1. Systemic Haemodynamics
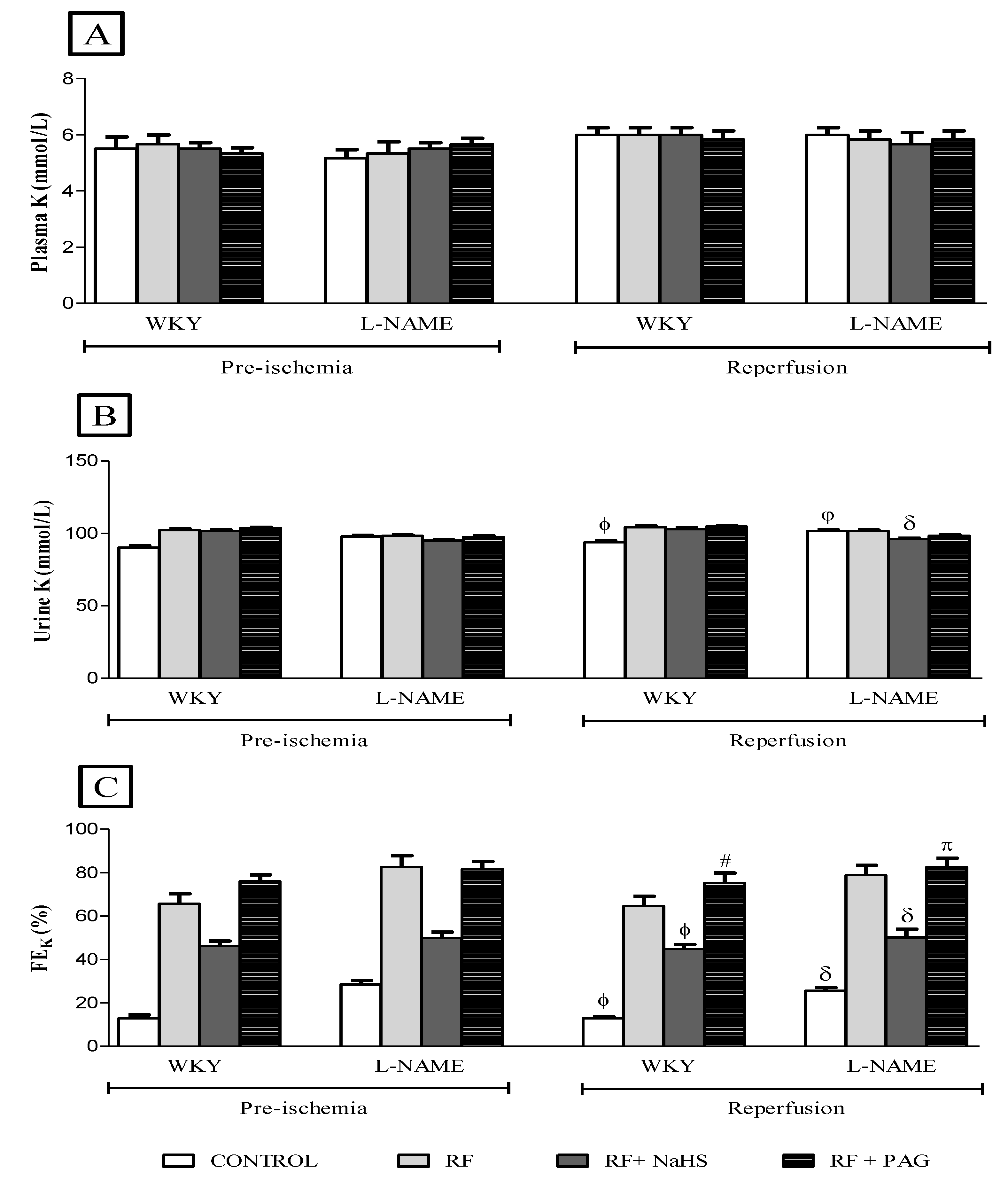
3.2. Renal Function Parameters
3.2.1. Plasma Concentration of H2S
3.2.2. Body Weight and Kidney Index on Day 36 (Acute Experiment day)
3.2.3. Renal Cortical Blood Perfusion in Pre-Ischemia and Reperfusion Phases
3.2.4. Renal Function Parameters in Pre-Ischemia and Reperfusion Phases
3.2.5. Oxidative Stress Markers in Pre-Ischemia and Reperfusion Phases
3.3. Concentration of ICAM-1 and NF-kB in Pre-Ischemia and Reperfusion Phases and Kidney Tissue
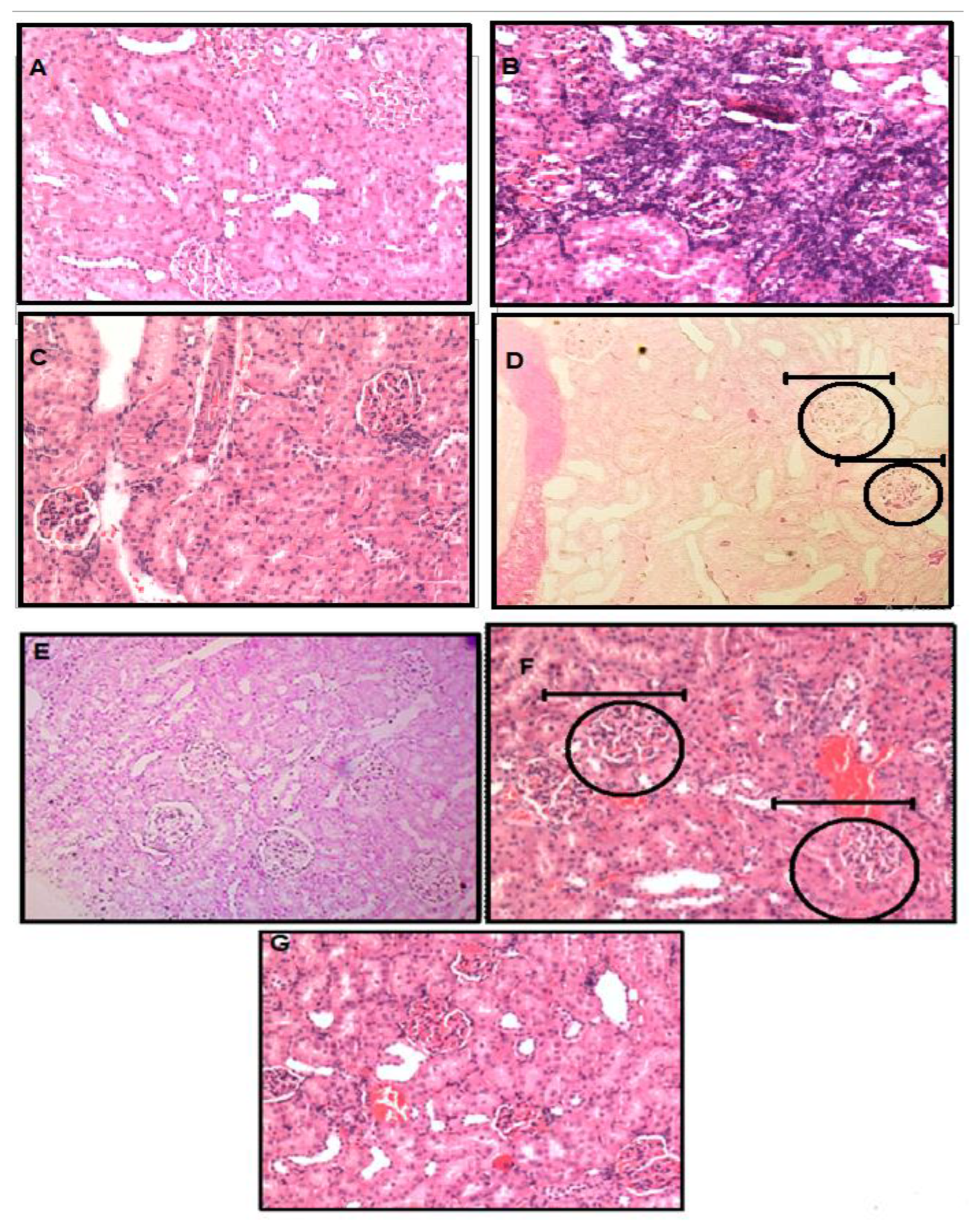
3.4. Histopathology of Kidney Tissue by Using Hematoxyllin and Eosin Staining
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lindner, J.R.; Coggins, M.P.; Kaul, S.; Klibanov, A.L.; Brandenburger, G.H.; Ley, K. Microbubble persistence in the microcirculation during ischemia/reperfusion and inflammation is caused by integrin-and complement-mediated adherence to activated leukocytes. Circulation 2000, 101, 668–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeschke, H. Mechanisms of Liver Injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am. J. Physiol.-Gastrointest. Liver Physiol. 2006, 290, G1083–G1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinhoff, G.; Behrend, M.; Schrader, B.; Duijvestijn, A.; Wonigeit, K. Expression patterns of leukocyte adhesion ligand molecules on human liver endothelia. Lack of ELAM-1 and CD62 inducibility on sinusoidal endothelia and distinct distribution of VCAM-1, ICAM-1, ICAM-2, and LFA-3. Am. J. Pathol. 1993, 142, 481. [Google Scholar] [PubMed]
- Vollmar, B.; Glasz, J.; Menger, M.D.; Messmer, K. Leukocytes contribute to hepatic ischemia/reperfusion injury via intercellular adhesion molecule-1-mediated venular adherence. Surgery 1995, 117, 195–200. [Google Scholar] [CrossRef]
- Collins, T.; Read, M.; Neish, A.; Whitley, M.; Thanos, D.; Maniatis, T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 1995, 9, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Nichols, T.C. NF-kB and reperfusion injury. Drug News Perspect 2004, 17, 99–104. [Google Scholar] [CrossRef]
- Chen, A.C.; Arany, P.R.; Huang, Y.-Y.; Tomkinson, E.M.; Sharma, S.K.; Kharkwal, G.B.; Saleem, T.; Mooney, D.; Yull, F.E.; Blackwell, T.S. Low-level laser therapy activates NF-kB via generation of reactive oxygen species in mouse embryonic fibroblasts. PLoS ONE 2011, 6, e22453. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.; Anwar, K.N.; True, A.L.; Malik, A.B. Thrombin-induced p65 homodimer binding to downstream NF-κB site of the promoter mediates endothelial ICAM-1 expression and neutrophil adhesion. J. Immunol. 1999, 162, 5466–5476. [Google Scholar]
- Singh, D.; Chander, V.; Chopra, K. Protective effect of catechin on ischemia-reperfusion-induced renal injury in rats. Pharm. Rep. 2005, 57, 70–76. [Google Scholar]
- Singh, D.; Chopra, K. The effect of naringin, a bioflavonoid on ischemia-reperfusion induced renal injury in rats. Pharmacol. Res. 2004, 50, 187–193. [Google Scholar] [CrossRef]
- Lee, M.; Schwab, C.; Yu, S.; McGeer, E.; McGeer, P.L. Astrocytes produce the antiinflammatory and neuroprotective agent hydrogen sulfide. Neurobiol. Aging 2009, 30, 1523–1534. [Google Scholar] [CrossRef]
- Szabó, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007, 6, 917–935. [Google Scholar] [CrossRef]
- Blackstone, E.; Morrison, M.; Roth, M.B. H2S induces a suspended animation–like state in mice. Science 2005, 308, 518. [Google Scholar] [CrossRef] [Green Version]
- Kuksis, M.; Smith, P.M.; Ferguson, A.V. Hydrogen sulfide regulates cardiovascular function by influencing the excitability of subfornical organ neurons. PLoS ONE 2014, 9, e105772. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, S.; Liu, H.; Zhang, B.; Zhao, Y.; Ma, K.; Zhao, D.; Wang, Q.; Ma, H.; Zhang, Z. Hydrogen Sulfide Prevents Hydrogen Peroxide-Induced Activation of Epithelial Sodium Channel through a PTEN/PI (3, 4, 5) P 3 Dependent Pathway. PLoS ONE 2013, 8, e64304. [Google Scholar]
- Stipanuk, M.H.; Beck, P.W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 1982, 206, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Swaroop, M.; Bradley, K.; Ohura, T.; Tahara, T.; Roper, M.D.; Rosenberg, L.; Kraus, J. Rat cystathionine beta-synthase. Gene organization and alternative splicing. J. Biol. Chem. 1992, 267, 11455–11461. [Google Scholar] [CrossRef]
- Tsai, C.-Y.; Peh, M.T.; Feng, W.; Dymock, B.W.; Moore, P.K. Hydrogen sulfide promotes adipogenesis in 3T3L1 cells. PLoS ONE 2015, 10, e0119511. [Google Scholar] [CrossRef]
- Zayachkivska, O.; Havryluk, O.; Hrycevych, N.; Bula, N.; Grushka, O.; Wallace, J.L. Cytoprotective effects of hydrogen sulfide in novel rat models of non-erosive esophagitis. PLoS ONE 2014, 9, e110688. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Sattar, M.A.; Rathore, H.A.; Abdulla, M.H.; Khan, S.A.; Azam, M.; Abdullah, N.A.; Johns, E.J. Up Regulation of cystathione γ lyase and Hydrogen Sulphide in the Myocardium Inhibits the Progression of Isoproterenol–Caffeine Induced Left Ventricular Hypertrophy in Wistar Kyoto Rats. PLoS ONE 2016, 11, e0150137. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.-D.; Wang, H.; Kho, S.-H.; Rinkiko, S.; Sheng, X.; Shen, H.-M.; Zhu, Y.-Z. Hydrogen sulfide protects HUVECs against hydrogen peroxide induced mitochondrial dysfunction and oxidative stress. PLoS ONE 2013, 8, e53147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, P.K.; Bhatia, M.; Moochhala, S. Hydrogen sulfide: From the smell of the past to the mediator of the future? Trends Pharmacol. Sci. 2003, 24, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Mikami, Y.; Kimura, Y.; Nagahara, N.; Kimura, H. Vascular endothelium expresses -mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J. Biochem. 2009, 146, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Ping, C.; Mok, Y.Y.; Ling, L.; Whiteman, M.; Bhatia, M.; Moore, P. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? Br. J. Pharmacol. 2006, 149, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine γ-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Zhong, G.; Chen, F.; Cheng, Y.; Tang, C.; Du, J. The role of hydrogen sulfide generation in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase. J. Hypertens. 2003, 21, 1879–1885. [Google Scholar] [CrossRef]
- Cheng, Y.; Ndisang, J.F.; Tang, G.; Cao, K.; Wang, R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am. J. Physiol.-Heart Circ. Physiol. 2004, 287, H2316–H2323. [Google Scholar] [CrossRef]
- Webb, G.D.; Lim, L.H.; Oh, V.M.; Yeo, S.B.; Cheong, Y.P.; Ali, M.Y.; El Oakley, R.; Lee, C.N.; Wong, P.S.; Caleb, M.G. Contractile and vasorelaxant effects of hydrogen sulfide and its biosynthesis in the human internal mammary artery. J. Pharmacol. Exp. Ther. 2008, 324, 876–882. [Google Scholar] [CrossRef]
- Al-Magableh, M.R.; Hart, J.L. Mechanism of vasorelaxation and role of endogenous hydrogen sulfide production in mouse aorta. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 383, 403–413. [Google Scholar] [CrossRef]
- Yang, C.; Yang, Z.; Zhang, M.; Dong, Q.; Wang, X.; Lan, A.; Zeng, F.; Chen, P.; Wang, C.; Feng, J. Hydrogen sulfide protects against chemical hypoxia-induced cytotoxicity and inflammation in HaCaT cells through inhibition of ROS/NF-κB/COX-2 pathway. PLoS ONE 2011, 6, e21971. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Olah, G.; Modis, K.; Coletta, C.; Kulp, G.; Gerö, D.; Szoleczky, P.; Chang, T.; Zhou, Z.; Wu, L. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc. Natl. Acad. Sci. USA 2011, 108, 13829–13834. [Google Scholar] [CrossRef] [Green Version]
- Lobb, I.; Zhu, J.; Liu, W.; Haig, A.; Lan, Z.; Sener, A. Hydrogen sulfide treatment ameliorates long-term renal dysfunction resulting from prolonged warm renal ischemia-reperfusion injury. Can. Urol. Assoc. J. 2014, 8, E413. [Google Scholar] [CrossRef] [Green Version]
- Lobb, I.; Mok, A.; Lan, Z.; Liu, W.; Garcia, B.; Sener, A. Supplemental hydrogen sulphide protects transplant kidney function and prolongs recipient survival after prolonged cold ischaemia–reperfusion injury by mitigating renal graft apoptosis and inflammation. BJU Int. 2012, 110, E1187–E1195. [Google Scholar] [CrossRef]
- Hashmi, S.F.; Rathore, H.A.; Sattar, M.A.; Johns, E.J.; Gan, C.-Y.; Chia, T.Y.; Ahmad, A. Hydrogen Sulphide Treatment Prevents Renal Ischemia-Reperfusion Injury by Inhibiting the Expression of ICAM-1 and NF-kB Concentration in Normotensive and Hypertensive Rats. Biomolecules 2021, 11, 1549. [Google Scholar] [CrossRef]
- Aminzadeh, M.A.; Vaziri, N.D. Downregulation of the renal and hepatic hydrogen sulfide (H2S)-producing enzymes and capacity in chronic kidney disease. Nephrol. Dial. Transplant. 2012, 27, 498–504. [Google Scholar] [CrossRef] [Green Version]
- Santos, N.; Catao, C.; Martins, N.; Curti, C.; Bianchi, M.; Santos, A. Cisplatin-induced nephrotoxicity is associated with oxidative stress, redox state unbalance, impairment of energetic metabolism and apoptosis in rat kidney mitochondria. Arch. Toxicol. 2007, 81, 495–504. [Google Scholar] [CrossRef]
- Niu, X.; Smith, C.W.; Kubes, P. Intracellular oxidative stress induced by nitric oxide synthesis inhibition increases endothelial cell adhesion to neutrophils. Circ. Res. 1994, 74, 1133–1140. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.H.; Sattar, M.A.; Abdullah, N.A.; Johns, E.J. 1B Adrenoceptors mediate adrenergically induced renal vasoconstrictions in rats with renal impairment. Acta Pharmacol. Sin. 2008, 29, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Yousef, M.I.; Saad, A.A.; El-Shennawy, L.K. Protective effect of grape seed proanthocyanidin extract against oxidative stress induced by cisplatin in rats. Food Chem. Toxicol. 2009, 47, 1176–1183. [Google Scholar] [CrossRef]
- Afkir, S.; Nguelefack, T.B.; Aziz, M.; Zoheir, J.; Cuisinaud, G.; Bnouham, M.; Mekhfi, H.; Legssyer, A.; Lahlou, S.; Ziyyat, A. Arbutus unedo prevents cardiovascular and morphological alterations in L-NAME-induced hypertensive rats: Part I: Cardiovascular and renal hemodynamic effects of Arbutus unedo in L-NAME-induced hypertensive rats. J. Ethnopharmacol. 2008, 116, 288–295. [Google Scholar] [CrossRef]
- Saravanakumar, M.; Raja, B. Veratric acid, a phenolic acid attenuates blood pressure and oxidative stress in L-NAME induced hypertensive rats. Eur. J. Pharmacol. 2011, 671, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Du, J.; Tang, C. The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem. Biophys. Res. Commun. 2004, 313, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Sattar, M.Z.; Rathore, H.A.; Akhtar, S.; Khan, M.A.; Hashmi, F.; Abdullah, N.; Johns, E.J. Impact of isoprenaline and caffeine on development of left ventricular hypertrophy and renal hemodynamic in wistar kyoto rats. Measurements 2012, 76, 1015–1026. [Google Scholar]
- Abdulla, M.H.; Sattar, M.A.; Abdullah, N.A.; Khan, M.A.H.; Abdallah, H.; Johns, E.J. Chronic treatment with losartan and carvedilol differentially modulates renal vascular responses to sympathomimetics compared to treatment with individual agents in normal Wistar Kyoto and spontaneously hypertensive rats. Eur. J. Pharmacol. 2009, 612, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Kiew, L.V.; Munavvar, A.S.; Law, C.H.; Azizan, A.N.; Nazarina, A.R.; Sidik, K.; Johns, E.J. Effect of antisense oligodeoxynucleotides for ICAM-1 on renal ischaemia–reperfusion injury in the anaesthetised rat. J. Physiol. 2004, 557, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.u.D.; Sattar, M.A.; Rathore, H.A.; Abdullah, M.H.; Tan, S.; Abdullah, N.A.; Johns, E.J. Exogenous hydrogen sulfide (H2S) reduces blood pressure and prevents the progression of diabetic nephropathy in spontaneously hypertensive rats. Ren. Fail. 2012, 34, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Seeling, H.; Wust, H. Colorimetric method for determination of creatinine. Arztl. Lab 1969, 15, 34. [Google Scholar]
- Brahmi, D.; Ayed, Y.; Hfaiedh, M.; Bouaziz, C.; Mansour, H.B.; Zourgui, L.; Bacha, H. Protective effect of cactus cladode extract against cisplatin induced oxidative stress, genotoxicity and apoptosis in balb/c mice: Combination with phytochemical composition. BMC Complement. Altern. Med. 2012, 12, 111. [Google Scholar] [CrossRef] [Green Version]
- Bell, D.; Zhao, Y.; McCoy, F.P.; Devine, A.B.; McDermott, B.J. Differential effects of an anti-oxidant intervention on cardiomyocyte expression of adrenomedullin and intermedin and their receptor components in chronic nitric oxide deficiency. Cell. Physiol. Biochem. 2008, 20, 269–282. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Hüttemann, M. Molecular mechanisms of ischemia–reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Al-Harbi, M.; Osman, A.-M.; Al-Gharably, N.; Al-Bekairi, A.; Al-Shabanah, O.; Sabah, D.; Raza, M. Effect of desferrioxamine on cisplatin-induced nephrotoxicity in normal rats. Chemotherapy 1995, 41, 448–454. [Google Scholar] [CrossRef]
- Tomita, H.; Egashira, K.; Kubo-Inoue, M.; Usui, M.; Koyanagi, M.; Shimokawa, H.; Takeya, M.; Yoshimura, T.; Takeshita, A. Inhibition of NO synthesis induces inflammatory changes and monocyte chemoattractant protein-1 expression in rat hearts and vessels. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1456–1464. [Google Scholar] [CrossRef] [Green Version]
- Jordan, J.E.; Zhao, Z.-Q.; Vinten-Johansen, J. The role of neutrophils in myocardial ischemia–reperfusion injury. Cardiovasc. Res. 1999, 43, 860–878. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, H.; Bautista, A.; Spolarics, Z.; Spitzer, J. Superoxide generation by neutrophils and Kupffer cells during in vivo reperfusion after hepatic ischemia in rats. J. Leukoc. Biol. 1992, 52, 377–382. [Google Scholar] [CrossRef]
- Mehrabi, A.; Mood, Z.A.; Sadeghi, M.; Schmied, B.; Müller, S.; Welsch, T.; Kuttymuratov, G.; Wente, M.; Weitz, J.; Zeier, M. Thymoglobulin and ischemia-reperfusion injury in kidney and liver transplantation. Nephrol. Dial. Transplant. 2007, 22, viii54–viii60. [Google Scholar] [CrossRef]
- Weitz, J.; Koch, M.; Mehrabi, A.; Schemmer, P.; Zeier, M.; Beimler, J.; Büchler, M.; Schmidt, J. Living-donor kidney transplantation: Risks of the donor–benefits of the recipient. Clin. Transplant. 2006, 20, 13–16. [Google Scholar] [CrossRef]
- Kim, S.-R.; Bae, Y.-H.; Bae, S.-K.; Choi, K.-S.; Yoon, K.-H.; Koo, T.H.; Jang, H.-O.; Yun, I.; Kim, K.-W.; Kwon, Y.-G. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-κB activation in endothelial cells. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2008, 1783, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Sattar, M.; Rathore, H.; Abdulla, M.; Ahmad, A.; Afzal, S.; Abdullah, N.; Johns, E. Renal denervation restores the baroreflex control of renal sympathetic nerve activity and heart rate in Wistar-Kyoto rats with cisplatin-induced renal failure. Acta Physiol. 2014, 210, 690–700. [Google Scholar] [CrossRef]
- Appenroth, D.; Frob, S.; Kersten, L.; Splinter, F.K.; Winnefeld, K. Protective effects of vitamin E and C on cisplatin nephrotoxicity in developing rats. Arch. Toxicol. 1997, 71, 677–683. [Google Scholar] [CrossRef]
- Sugihara, K.; Gemba, M. Modification of cisplatin toxicity by antioxidants. Jpn. J. Pharmacol. 1986, 40, 353–355. [Google Scholar] [CrossRef] [Green Version]
- Ahangarpour, A.; Fard, A.A.; Gharibnaseri, M.K.; Jalali, T.; Rashidi, I. Hydrogen sulfide ameliorates the kidney dysfunction and damage in cisplatin-induced nephrotoxicity in rat. In Veterinary Research Forum: An International Quarterly Journal; Faculty of Veterinary Medicine, Urmia University: Urmia, Iran, 2014; p. 121. [Google Scholar]
- Sanchez-Gonzalez, P.D.; Lopez-Hernandez, F.J.; Lopez-Novoa, J.M.; Morales, A.I. An integrative view of the pathophysiological events leading to cisplatin nephrotoxicity. Crit. Rev. Toxicol. 2011, 41, 803–821. [Google Scholar] [CrossRef] [PubMed]
- Snijder, P.M.; Frenay, A.-R.S.; Koning, A.M.; Bachtler, M.; Pasch, A.; Kwakernaak, A.J.; van den Berg, E.; Bos, E.M.; Hillebrands, J.-L.; Navis, G. Sodium thiosulfate attenuates angiotensin II-induced hypertension, proteinuria and renal damage. Nitric Oxide 2014, 42, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Chia, T.Y.; Sattar, M.A.; Abdulla, M.H.; Rathore, H.A.; Ahmad, F.u.D.; Kaur, G.; Abdullah, N.A.; Johns, E.J. The effects of tempol on renal function and hemodynamics in cyclosporine-induced renal insufficiency rats. Ren. Fail. 2013, 35, 978–988. [Google Scholar] [CrossRef]
- Zicha, J.; Dobesova, Z.; Kunes, J. Antihypertensive mechanisms of chronic captopril or N-acetylcysteine treatment in L-NAME hypertensive rats. Hypertens. Res. 2006, 29, 1021–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, D.; Palmer, R.; Schulz, R.; Hodson, H.; Moncada, S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br. J. Pharmacol. 1990, 101, 746–752. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef] [Green Version]
- Leibbrandt, M.E.; Wolfgang, G.H.; Metz, A.L.; Ozobia, A.A.; Haskins, J.R. Critical subcellular targets of cisplatin and related platinum analogs in rat renal proximal tubule cells. Kidney Int. 1995, 48, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Yatsu, T.; Aoki, M.; Inagaki, O. Preventive effect of zelandopam, a dopamine D1 receptor agonist, on cisplatin-induced acute renal failure in rats. Eur. J. Pharmacol. 2003, 461, 191–195. [Google Scholar] [CrossRef]
- Xavier, F.; Magalhães, A.; Gontijo, J. Effect of inhibition of nitric oxide synthase on blood pressure and renal sodium handling in renal denervated rats. Braz. J. Med. Biol. Res. 2000, 33, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Muchant, D.; Beierwaltes, W.H.; Racusen, L.; Baylis, C. Evolution of chronic nitric oxide inhibition hypertension Relationship to renal function. Hypertension 1998, 31, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Brahmbhatt, S.; Gupta, A.; Sharma, A.C. Duration of streptozotocin-induced diabetes differentially affects p 38-mitogen-activated protein kinase(MAPK) phosphorylation in renal and vascular dysfunction. Cardiovasc. Diabetol. 2005, 4, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Chia, T.Y.; Sattar, M.A.; Rathore, H.A.; Abdullah, M.H.; Singh, C.; Kaur, G.; Ahmad, D.; Ud, F.; Ibraheem, Z.O.; Li, K.J. Superoxide Dismutase Mimetic Tempol Normalized the Blood Pressure and Renal Functions in L-NAME Induced Hypertension Rats: The Role of Oxidative Stress. Int. J. Pharm. Sci. Rev. Res. 2013, 18, 61–71. [Google Scholar]
- Kang, D.G.; Sohn, E.J.; Lee, Y.M.; Lee, A.S.; Han, J.H.; Kim, T.Y.; Lee, H.S. Effects of bulbus Fritillaria water extract on blood pressure and renal functions in the L-NAME-induced hypertensive rats. J. Ethnopharmacol. 2004, 91, 51–56. [Google Scholar] [CrossRef]
- Perna, A.F.; Ingrosso, D. Low hydrogen sulphide and chronic kidney disease: A dangerous liaison. Nephrol. Dial. Transplant. 2012, 27, 486–493. [Google Scholar] [CrossRef]
- Winston, J.A.; Safirstein, R. Reduced renal blood flow in early cisplatin-induced acute renal failure in the rat. Am. J. Physiol.-Ren. Physiol. 1985, 249, F490–F496. [Google Scholar] [CrossRef]
- Ladefoged, J. Increase in renal blood flow in acute renal failure following intra-arterial infusion of acetylcholine. Scand. J. Clin. Lab. Investig. 1977, 37, 709–712. [Google Scholar] [CrossRef]
- Xia, M.; Chen, L.; Muh, R.W.; Li, P.-L.; Li, N. Production and actions of hydrogen sulfide, a novel gaseous bioactive substance, in the kidneys. J. Pharmacol. Exp. Ther. 2009, 329, 1056–1062. [Google Scholar] [CrossRef]
- Awad, A.S.; Ye, H.; Huang, L.; Li, L.; Foss, F.W.; Macdonald, T.L.; Lynch, K.R.; Okusa, M.D. Selective sphingosine 1-phosphate 1 receptor activation reduces ischemia-reperfusion injury in mouse kidney. Am. J. Physiol.-Ren. Physiol. 2006, 290, F1516–F1524. [Google Scholar] [CrossRef] [Green Version]
- Saleh, S.; El-Demerdash, E. Protective Effects of L-Arginine against Cisplatin-Induced Renal Oxidative Stress and Toxicity: Role of Nitric Oxide. Basic Clin. Pharmacol. Toxicol. 2005, 97, 91–97. [Google Scholar] [CrossRef]
- Yoshida, I.; Bengal, R.; Torres, V.E. Gender-dependent effect of L-NAME on polycystic kidney disease in Han: SPRD rats. Am. J. Kidney Dis. 2000, 35, 930–936. [Google Scholar] [CrossRef]
- Basile, D.P.; Donohoe, D.; Roethe, K.; Osborn, J.L. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am. J. Physiol.-Ren. Physiol. 2001, 281, F887–F899. [Google Scholar] [CrossRef]
- Bagheri, F.; Gol, A.; Dabiri, S.; Javadi, A. Preventive effect of garlic juice on renal reperfusion injury. Iran. J. Kidney Dis. 2011, 5, 194. [Google Scholar] [PubMed]
- Kim, J.; Jang, H.-S.; Park, K.M. Reactive oxygen species generated by renal ischemia and reperfusion trigger protection against subsequent renal ischemia and reperfusion injury in mice. Am. J. Physiol.-Ren. Physiol. 2010, 298, F158–F166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Prathapasinghe, G.; Wu, N.; Hwang, S.-Y.; Siow, Y.L.; Karmin, O. Ischemia-reperfusion reduces cystathionine-β-synthase-mediated hydrogen sulfide generation in the kidney. Am. J. Physiol.-Ren. Physiol. 2009, 297, F27–F35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.S.; Sharples, E.J.; Cuzzocrea, S.; Chatterjee, P.K.; Britti, D.; Yaqoob, M.M.; Thiemermann, C. Pretreatment with EPO reduces the injury and dysfunction caused by ischemia/reperfusion in the mouse kidney in vivo. Kidney Int. 2004, 66, 983–989. [Google Scholar] [CrossRef] [Green Version]
- Şener, G.; Şehirli, Ö.; Velioğlu-Öğünç, A.; Çetinel, Ş.; Gedik, N.; Caner, M.; Sakarcan, A.; Yeğen, B.Ç. Montelukast protects against renal ischemia/reperfusion injury in rats. Pharmacol. Res. 2006, 54, 65–71. [Google Scholar] [CrossRef]
- Hosseinzadeh, H.; Sadeghnia, H.R.; Ziaee, T.; Danaee, A. Protective effect of aqueous saffron extract (Crocus sativus L.) and crocin, its active constituent, on renal ischemia-reperfusion-induced oxidative damage in rats. J. Pharm. Pharm. Sci. 2005, 8, 387–393. [Google Scholar]
- Nakmareong, S.; Kukongviriyapan, U.; Pakdeechote, P.; Donpunha, W.; Kukongviriyapan, V.; Kongyingyoes, B.; Sompamit, K.; Phisalaphong, C. Antioxidant and vascular protective effects of curcumin and tetrahydrocurcumin in rats with L-NAME-induced hypertension. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 383, 519–529. [Google Scholar] [CrossRef]
- Simko, F.; Pechanova, O.; Pelouch, V.; Krajcirovicova, K.; Celec, P.; Palffy, R.; Bednarova, K.; Vrankova, S.; Adamcova, M.; Paulis, L. Continuous light and L-NAME-induced left ventricular remodelling: Different protection with melatonin and captopril. J. Hypertens. 2010, 28, S13–S18. [Google Scholar] [CrossRef]
- Matsushima, H.; Yonemura, K.; Ohishi, K.; Hishida, A. The role of oxygen free radicals in cisplatin-induced acute renal failure in rats. J. Lab. Clin. Med. 1998, 131, 518–526. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.V.; Walker, P.D. Evidence suggesting a role for hydroxyl radical in glycerol-induced acute renal failure. Am. J. Physiol.-Ren. Physiol. 1988, 255, F438–F443. [Google Scholar] [CrossRef]
- Çetin, R.; Devrim, E.; Kılıçoğlu, B.; Avcı, A.; Çandır, Ö.; Durak, İ. Cisplatin impairs antioxidant system and causes oxidation in rat kidney tissues: Possible protective roles of natural antioxidant foods. J. Appl. Toxicol. 2006, 26, 42–46. [Google Scholar] [CrossRef]
- Feng, L.; Ke, N.; Cheng, F.; Guo, Y.; Li, S.; Li, Q.; Li, Y. The protective mechanism of ligustrazine against renal ischemia/reperfusion injury. J. Surg. Res. 2011, 166, 298–305. [Google Scholar] [CrossRef]
- Aydogdu, N.; Atmaca, G.; Yalcin, O.; Taskiran, R.; Tastekin, E.; Kaymak, K. Protective Effects of L-Carnitine on Myoglobinuric Acute Renal Failure in Rats. Clin. Exp. Pharmacol. Physiol. 2006, 33, 119–124. [Google Scholar] [CrossRef]
- Whiteman, M.; Armstrong, J.S.; Chu, S.H.; Jia-Ling, S.; Wong, B.S.; Cheung, N.S.; Halliwell, B.; Moore, P.K. The novel neuromodulator hydrogen sulfide: An endogenous peroxynitrite ‘scavenger’? J. Neurochem. 2004, 90, 765–768. [Google Scholar] [CrossRef]
- Yan, S.-K.; Chang, T.; Wang, H.; Wu, L.; Wang, R.; Meng, Q.H. Effects of hydrogen sulfide on homocysteine-induced oxidative stress in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2006, 351, 485–491. [Google Scholar] [CrossRef]
- Bayrak, O.; Uz, E.; Bayrak, R.; Turgut, F.; Atmaca, A.F.; Sahin, S.; Yıldırım, M.E.; Kaya, A.; Cimentepe, E.; Akcay, A. Curcumin protects against ischemia/reperfusion injury in rat kidneys. World J. Urol. 2008, 26, 285–291. [Google Scholar] [CrossRef]
- Nakano, S.; GEMBA, M. Potentiation of cisplatin-induced lipid peroxidation in kidney cortical slices by glutathione depletion. Jpn. J. Pharmacol. 1989, 50, 87–92. [Google Scholar] [CrossRef]
- Kimura, Y.; Goto, Y.I.; Kimura, H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal. 2010, 12, 1–13. [Google Scholar] [CrossRef]
- Kimura, Y.; Kimura, H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004, 18, 1165–1167. [Google Scholar] [CrossRef]
- Carden, D.L.; Smith, J.K.; Korthuis, R.J. Neutrophil-mediated microvascular dysfunction in postischemic canine skeletal muscle. Role of granulocyte adherence. Circ. Res. 1990, 66, 1436–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekido, N.; Mukaida, N.; Harada, A.; Nakanishi, I.; Watanabe, Y.; Matsushima, K. Prevention of lung reperfusion injury in rabbits by a monoclonal antibody against interleukin-8. Nature 1993, 365, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Schreck, R.; Albermann, K.; Baeuerle, P.A. Nuclear factor kB: An oxidative stress-responsive transcription factor of eukaryotic cells (a review). Free Radic. Res. Commun. 1992, 17, 221–237. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Names of Groups | Day 0 | Day 21 | Day 35 |
|---|---|---|---|---|
| Body Weight | WKY-CONTROL | 236 ± 2 | 272 ± 4 ψ | 316.0 ± 8.9 ψ ϕ δ |
| WKY-RF | 240 ± 6 | 277 ± 10.0 ψ | 282.5 ± 17.2 ψ | |
| WKY-RF+NaHS | 236 ± 5 | 279 ± 12 ψ | 276.8 ± 6.4 ψ | |
| WKY-RF+PAG | 231 ± 6 | 272 ± 7 ψ | 267 ± 5 ψ | |
| L-NAME-CONTROL | 233 ± 5 | 260.7 ± 4.1 ψ | 284 ± 6 ψ ϕ φ π | |
| L-NAME-RF | 232 ± 4 | 262.2 ± 4.2 ψ | 259 ± 7 ψ δ | |
| L-NAME-RF+NaHS | 228 ± 5 | 266.5 ± 2.2 ψ | 273 ± 3ψ φ | |
| L-NAME-RF+PAG | 234 ± 6 | 260 ± 5 ψ | 261 ± 8 ψ | |
| Water Intake | WKY-CONTROL | 50± 6 | 51.3 ± 5.6 | 51.6 ± 6.1 |
| WKY-RF | 43 ± 5 | 42 ± 6 | 56 ± 11 | |
| WKY-RF+NaHS | 44 ± 5 | 46.6 ± 3.0 | 52 ± 7 | |
| WKY-RF+PAG | 41 ± 8 | 39.6 ± 5.9 | 53 ± 6 | |
| L-NAME-CONTROL | 42 ± 5 | 47.0 ± 4.8 | 52.5 ± 9 | |
| L-NAME-RF | 43 ± 5 | 42.1 ± 5.5 | 56 ± 11 | |
| L-NAME-RF+NaHS | 43 ± 6 | 42.5 ± 4.7 | 48 ±5 | |
| L-NAME-RF+PAG | 45 ±7 | 47.3 ± 4.9 | 52 ± 19 | |
| Urinary Out Put | WKY-CONTROL | 21 ± 3 | 21 ± 3 | 20 ± 4 |
| WKY-RF | 16 ± 2 | 16 ± 2 | 31 ± 8 ψ ϕ | |
| WKY-RF+NaHS | 16 ± 2 | 16 ± 5 | 25 ± 3 ϕ | |
| WKY-RF+PAG | 21 ± 4 | 19 ± 3 | 28 ± 5 | |
| L-NAME-CONTROL | 19.8 ± 6.9 | 17 ± 8 | 19 ± 8 φ | |
| L-NAME-RF | 14.8 ± 2.1 | 18± 3 | 28.6 ± 6.2 ψ ϕ | |
| L-NAME-RF+NaHS | 14 ± 2 | 14± 2 | 19 ± 2 φ | |
| L-NAME-RF+PAG | 14.1 ± 1.4 | 17.6 ± 2.4 | 28 ± 4 ψ ϕ | |
| Urine flow rate | WKY-CONTROL | 6 ± 0.8 | 5 ± 0.7 | 4 ± 0.7 δ |
| WKY-RF | 5 ± 0.6 | 4 ± 0.5 | 8 ± 3 ψ ϕ | |
| WKY-RF+NaHS | 5 ± 0.5 | 4 ± 1.0 | 6± 0.8 | |
| WKY-RF+PAG | 6.24 ± 1.5 | 4.90 ± 0.8 | 7 ± 1 | |
| L-NAME-CONTROL | 6 ± 2 | 4 ± 2 | 5 ± 2 φ | |
| L-NAME-RF | 4 ± 0.6 | 5 ± 0.7 | 8 ± 2 ψ ϕ | |
| L-NAME-RF+NaHS | 4 ± 0.7 | 4 ± 0.5 | 5 ± 0.5 φ | |
| L-NAME-RF+PAG | 4 ± 0.3 | 5 ± 0.5 | 7 ± 1 ψ ϕ |
| Parameters | Groups | Day 0 | Day 21 | Day 35 |
|---|---|---|---|---|
| Systolic blood pressure (SBP) | WKY-CONTROL | 119 ± 7 | 121 ± 1 | 118 ± 4 |
| WKY-RF | 115 ± 3 | 119 ± 4 | 116 ± 4 | |
| WKY-RF+NaHS | 118 ± 4 | 116 ± 2 | 116 ± 3 | |
| WKY-RF+PAG | 119 ± 3 | 116.5 ± 3.2 | 118.2 ± 2.2 | |
| L-NAME-CONTROL | 121 ± 3 | 174 ± 10 ψ | 200 ± 12 ψ ϕ π | |
| L-NAME-RF | 119 ± 4 | 162 ± 6 ψ | 193 ± 6 ψ ϕ δ | |
| L-NAME-RF+NaHS | 117 ± 3 | 139 ± 3 ψ | 176 ± 70 ψ ϕ φ | |
| L-NAME-RF+PAG | 115 ± 5 | 159 ± 4 ψ | 196 ± 7 ψ ϕ | |
| Diastolic blood pressure (DBP) | WKY-CONTROL | 89 ± 5 | 88 ± 2 | 89 ± 1 |
| WKY-RF | 89± 2 | 89 ± 2 | 89 ± 2 | |
| WKY-RF+NaHS | 91 ± 2 | 89 ± 3 | 88 ± 2 | |
| WKY-RF+PAG | 88 ± 1 | 88 ± 2 | 89 ± 1 | |
| L-NAME-CONTROL | 91 ± 2 | 128 ± 6 ψ | 156 ± 6 ψ ϕ π | |
| L-NAME-RF | 90 ± 2 | 133 ± 4 ψ | 159 ± 5 ψ ϕ δ | |
| L-NAME-RF+NaHS | 88 ± 1 | 110 ± 2 ψ | 132 ± 4 ψ ϕ φ | |
| L-NAME-RF+PAG | 88 ± 2 | 128 ± 12 ψ | 159 ± 4 ψ ϕ | |
| Mean arterial pressure (MAP) | WKY-CONTROL | 99 ± 4 | 99 ± 1 | 98 ± 2 |
| WKY-RF | 97 ± 2 | 99 ± 0.5 | 97 ± 1 | |
| WKY-RF+NaHS | 99 ± 1 | 98 ± 2 | 96 ± 2 | |
| WKY-RF+PAG | 98± 1.0 | 98 ± 1 | 9.8 ± 1 | |
| L-NAME-CONTROL | 101 ± 2 | 143 ± 3 ψ | 171 ± 5 ψ ϕ π | |
| L-NAME-RF | 99 ± 2 | 142 ± 4 ψ | 172 ± 4 ψ ϕ δ | |
| L-NAME-RF+NaHS | 98± 1 | 119 ± 2 ψ | 146.0 ± 4 ψ ϕ φ | |
| L-NAME-RF+PAG | 97 ± 2 | 141 ± ψ | 173 ± 4 ψ ϕ | |
| Heart rate (HR) | WKY-CONTROL | 325 ± 10 | 322.7 ± 15 | 309 ± 14 |
| WKY-RF | 316± 9 | 322 ± 10 | 308 ± 10 | |
| WKY-RF+NaHS | 321± 30 | 303 ± 18 | 320 ± 33 | |
| WKY-RF+PAG | 315± 8 | 304 ± 16 | 306 ± 16 | |
| L-NAME-CONTROL | 314± 11 | 371 ± 12 ψ | 412 ± 14 ψ ϕ π | |
| L-NAME-RF | 313 ± 14 | 373 ± 11 ψ | 419 ± 23ψ ϕ δ | |
| L-NAME-RF+NaHS | 314 ± 7 | 332 ± 9 | 402 ± 16 ψ ϕ | |
| L-NAME-RF+PAG | 302 ± 18 | 385 ± 5 ψ | 428 ± 20 ψ ϕ |
| Parameters | Groups | Day 0 | Day 21 | Day 35 |
|---|---|---|---|---|
| Sodium in plasma | WKY-CONTROL | 129 ± 1.9 | 131 ± 2.3 | 129 ± 3.4 δ |
| WKY-RF | 122 ± 3.3 | 125 ± 2.8 | 105 ± 2.8 ψ ϕ | |
| WKY-RF+NaHS | 131 ± 2.6 | 129 ± 2.4 | 114 ± 2.6 ψ ϕ δ | |
| WKY-RF+PAG | 125 ± 3.8 | 127 ± 3.0 | 104 ± 2.9 ψ ϕ | |
| L-NAME-CONTROL | 127 ± 1.4 | 121 ± 1.4 ψ | 115 ± 1.4 ψ ϕ φ π | |
| L-NAME-RF | 128 ± 1.5 | 122 ± 1.7 ψ | 102 ± 1.9 ψ ϕ | |
| L-NAME-RF+NaHS | 125 ± 1.4 | 121 ± 1.5 | 107 ± 1.4 ψ ϕ | |
| L-NAME-RF+PAG | 129 ± 1.2 | 123 ± 1.3 ψ | 100 ± 1.0 ψ ϕ | |
| Potassium in urine | WKY-CONTROL | 112 ± 2.0 | 113 ± 2.2 | 112 ± 2.4 δ |
| WKY-RF | 111 ± 2.0 | 113 ± 2.0 | 124 ± 3.7 ψ ϕ | |
| WKY-RF+NaHS | 114 ± 2.6 | 112 ± 2.4 | 125 ± 3.8 ψ ϕ | |
| WKY-RF+PAG | 114 ± 2 | 112 ± 2.2 | 125 ± 4.1 ψ ϕ | |
| L-NAME-CONTROL | 114.2 ± 1.4 | 118 ± 1.5 | 122 ± 1.6 ψ φ π | |
| L-NAME-RF | 114 ± 1.0 | 118 ± 1.3 | 129 ± 1.7 ψ ϕ δ | |
| L-NAME-RF+NaHS | 113 ± 1.6 | 117 ± 1.3 | 126 ± 2.1 ψ ϕ | |
| L-NAME-RF+PAG | 112 ± 1.2 | 116 ± 1.4 | 130 ± 1.7 ψ ϕ | |
| Potassium in plasma | WKY-CONTROL | 5.33 ± 0.5 | 5.66 ± 1.2 | 5.50 ± 1.0 |
| WKY-RF | 5.16 ± 0.7 | 5.50 ± 0.8 | 5.66 ± 0.8 | |
| WKY-RF+NaHS | 5.33 ± 1.0 | 5.50 ± 0.5 | 5.33 ± 0.5 | |
| WKY-RF+PAG | 5.50 ± 0.8 | 5.83 ± 0.7 | 5.33 ± 0.5 | |
| L-NAME-CONTROL | 5.66 ± 0.8 | 5.33 ± 1.0 | 5.50 ± 1.0 | |
| L-NAME-RF | 5.66 ± 0.8 | 5.83 ± 0.9 | 5.66 ± 0.8 | |
| L-NAME-RF+NaHS | 5.33 ± 1.0 | 5.50 ± 0.5 | 5.16 ± 0.7 | |
| L-NAME-RF+PAG | 5.33 ± 0.5 | 5.83 ± 0.7 | 5.50 ± 1.0 | |
| Potassium in urine | WKY-CONTROL | 88.0 ± 1.4 | 90.3 ± 1.7 | 88.3 ± 2.5 δ |
| WKY-RF | 81.5 ± 2.4 | 83.6 ± 2.1 | 103.3 ± 2.1 ψ ϕ | |
| WKY-RF+NaHS | 79.6 ± 2.5 | 82.6 ± 2.5 | 99.6 ± 2.5 ψ ϕ | |
| WKY-RF+PAG | 84.3 ± 1.8 | 87.5 ± 1.8 | 105.5 ± 1.8 ψ ϕ | |
| L-NAME-CONTROL | 87.0 ± 1.4 | 92.3 ± 1.5 ψ | 98.5 ± 1.5 ψ ϕ π | |
| L-NAME-RF | 85.8 ± 1.1 | 91.8 ± 1.1 ψ | 97.6 ± 1.3 ψ ϕ δ | |
| L-NAME-RF+NaHS | 83.5 ± 1.8 | 87.0 ± 1.4 | 93.1 ± 1.6 ψ ϕ φ | |
| L-NAME-RF+PAG | 86.1 ± 1.1 | 92.0 ± 2.0 ψ | 98.0 ± 3.0 ψ ϕ | |
| Fractional excretion of sodium (FENa) | WKY-CONTROL | 0.68 ± 0.04 | 0.62 ± 0.09 | 0.70 ± 0.15 δ |
| WKY-RF | 0.73 ± 0.07 | 0.69 ± 0.12 | 4.25 ± 0.34 ψ ϕ | |
| WKY-RF+NaHS | 0.64 ± 0.09 | 0.61 ± 0.17 | 2.76 ± 0.18 ψ ϕ δ | |
| WKY-RF+PAG | 0.66 ± 0.02 | 0.64 ± 0.01 | 4.44 ± 0.29 ψ ϕ | |
| L-NAME-CONTROL | 0.69 ± 0.01 | 0.97 ± 0.03 | 1.55 ± 0.04 ψ ϕ φπ | |
| L-NAME-RF | 0.68 ± 0.04 | 1.01 ± 0.04 | 5.78 ± 0.32 ψ ϕ δ | |
| L-NAME-RF+NaHS | 0.69 ± 0.06 | 0.91 ± 0.06 | 3.45 ± 0.15 ψ ϕ φ | |
| L-NAME-RF+PAG | 0.66 ± 0.05 | 0.97 ± 0.03 | 6.18 ± 0.29 ψ ϕ | |
| Fractional excretion of potassium (FEK) | WKY-CONTROL | 12.8 ± 1.1 | 12.7 ± 4.7 | 13.3 ± 2.8 δ |
| WKY-RF | 13.1 ± 3.2 | 11.7 ± 1.2 | 66.4 ± 7.5 ψ ϕ | |
| WKY-RF+NaHS | 11.3 ± 2.8 | 10.5 ± 2.4 | 47.5 ± 6.2 ψ ϕ δ | |
| WKY-RF+PAG | 11.2 ± 1.3 | 11.1 ± 1.4 | 73.2 ± 5.7 ψ ϕ | |
| L-NAME-CONTROL | 12.5 ± 1.0 | 17.6 ± 2.8 | 26.4 ± 4.5 ψ φ π | |
| L-NAME-RF | 11.8 ± 1.8 | 16.8 ± 2.8 | 80.1 ± 10.9 ψ ϕ δ | |
| L-NAME-RF+NaHS | 12.4 ± 2.8 | 15.1 ± 2.1 | 53.3 ± 6.3 ψ ϕ φ | |
| L-NAME-RF+PAG | 12.3 ± 1.4 | 17.4 ± 2.3 | 87.2 ± 17.2 ψ ϕ | |
| Urinary sodium volume (UNaV) | WKY-CONTROL | 0.096 ± 0.013 | 0.098 ± 0.013 | 0.095 ± 0.016 δ |
| WKY-RF | 0.075 ± 0.011 | 0.074 ± 0.009 | 0.161 ± 0.046 ψ ϕ | |
| WKY-RF+NaHS | 0.075 ± 0.010 | 0.072 ± 0.021 | 0.130 ± 0.020 ψ ϕ | |
| WKY-RF+PAG | 0.098 ± 0.023 | 0.090 ± 0.014 | 0.147 ± 0.028 ψ ϕ | |
| L-NAME-CONTROL | 0.094 ± 0.033 | 0.083 ± 0.037 | 0.095 ± 0.040 φ | |
| L-NAME-RF | 0.070 ± 0.009 | 0.087 ± 0.013 | 0.154 ± 0.032 ψϕ | |
| L-NAME-RF+NaHS | 0.066 ± 0.011 | 0.066 ± 0.010 | 0.098 ± 0.011 φ | |
| L-NAME-RF+PAG | 0.066 ± 0.006 | 0.085 ± 0.011 | 0.153 ± 0.026 ψ ϕ | |
| Urinary potassium volume (UkV) | WKY-CONTROL | 0.104 ± 0.017 | 0.105 ± 0.016 | 0.107 ± 0.021 |
| WKY-RF | 0.122 ± 0.021 | 0.129 ± 0.015 | 0.084 ± 0.021 | |
| WKY-RF+NaHS | 0.123 ± 0.020 | 0.139 ± 0.044 | 0.097 ± 0.014 | |
| WKY-RF+PAG | 0.103 ± 0.029 | 0.112 ± 0.018 | 0.092 ± 0.016 | |
| L-NAME-CONTROL | 0.115 ± 0.033 | 0.152 ± 0.057 | 0.145 ± 0.050 φ | |
| L-NAME-RF | 0.141 ± 0.020 | 0.127 ± 0.020 | 0.085 ± 0.020 ψ | |
| L-NAME-RF+NaHS | 0.146 ± 0.043 | 0.155 ± 0.022 | 0.121 ± 0.014 | |
| L-NAME-RF+PAG | 0.147 ± 0.017 | 0.127 ± 0.018 | 0.085 ± 0.015 ψ | |
| Na:K ratio | WKY-CONTROL | 1.27 ± 0.02 | 1.25 ± 0.02 | 1.27 ± 0.03 |
| WKY-RF | 1.36 ± 0.05 | 1.35 ± 0.04 | 1.20 ± 0.04 ψ ϕ | |
| WKY-RF+NaHS | 1.43 ± 0.04 | 1.35 ± 0.03 | 1.25 ± 0.05 ψ ϕ | |
| WKY-RF+PAG | 1.35 ± 0.02 | 1.28 ± 0.04 | 1.18 ± 0.02 ψ ϕ | |
| L-NAME-CONTROL | 1.31 ± 0.02 | 1.28 ± 0.02 | 1.24 ± 0.01 φ | |
| L-NAME-RF | 1.33 ± 0.02 | 1.28 ± 0.01 | 1.32 ± 0.01 δ | |
| L-NAME-RF+NaHS | 1.36 ± 0.04 | 1.35 ± 0.03 | 1.36 ± 0.04 | |
| L-NAME-RF+PAG | 1.30 ± 0.03 | 1.27 ± 0.03 | 1.33 ± 0.04 | |
| Creatinine In plasma | WKY-CONTROL | 0.84 ± 0.03 | 0.74 ± 0.13 | 0.82 ± 0.17 δ |
| WKY-RF | 0.85 ± 0.10 | 0.83 ± 0.12 | 2.21 ± 0.13 ψ ϕ | |
| WKY-RF+NaHS | 0.79 ± 0.09 | 0.78 ± 0.19 | 1.82 ± 0.08 ψ ϕ δ | |
| WKY-RF+PAG | 0.79 ± 0.03 | 0.81 ± 0.04 | 2.24 ± 0.02 ψ ϕ | |
| L-NAME-CONTROL | 0.85 ± 0.02 | 0.96 ± 0.02 | 1.25 ± 0.19 ψ ϕ φ π | |
| L-NAME-RF | 0.84 ± 0.05 | 0.99 ± 0.04 | 2.50 ± 0.04 ψ ϕ δ | |
| L-NAME-RF+NaHS | 0.81 ± 0.07 | 0.93 ± 0.05 | 1.98 ± 0.08 ψ ϕ φ | |
| L-NAME-RF+PAG | 0.84 ± 0.05 | 1.01 ± 0.06 | 2.55 ± 0.05 ψ ϕ | |
| Creatinine in urine | WKY-CONTROL | 106.4 ± 3.8 | 103.6 ± 3.2 | 101.8 ± 3.6 δ |
| WKY-RF | 105.4 ± 4.3 | 108.2 ± 3.1 | 61.7 ± 4.0 ψ ϕ | |
| WKY-RF+NaHS | 108.8 ± 5.3 | 111.5 ± 5.9 | 72.3 ± 6.2 ψ ϕ δ | |
| WKY-RF+PAG | 109.6 ± 2.5 | 111.1 ± 2.9 | 61.0 ± 2.0 ψ ϕ | |
| L-NAME-CONTROL | 110.7 ± 1.8 | 96.2 ± 3.0 ψ | 85.4 ± 2.8 ψ ϕ φ π | |
| L-NAME-RF | 110.6 ± 2.2 | 94.9 ± 2.8 ψ | 54.6 ± 2.1 ψ ϕ δ | |
| L-NAME-RF+NaHS | 105.9 ± 1.2 | 99.3 ± 1.6 | 68.0 ± 2.0 ψ ϕ φ | |
| L-NAME-RF+PAG | 111.9 ± 1.9 | 93.4 ± 1.2 ψ | 53.7 ± 1.6 ψ ϕ | |
| Creatinine clearance | WKY-CONTROL | 7.68 ± 1.38 | 7.62 ± 0.98 | 5.80 ± 0.64 δ |
| WKY-RF | 5.94 ± 1.25 | 5.26 ± 1.33 | 2.15 ± 0.72 ψ ϕ | |
| WKY-RF+NaHS | 6.36 ± 0.58 | 5.71 ± 1.90 | 2.48 ± 0.45 ψ ϕ | |
| WKY-RF+PAG | 8.58 ± 2.00 | 6.72 ± 1.22 | 1.99 ± 0.37 ψ ϕ | |
| L-NAME-CONTROL | 6.71 ± 1.71 | 4.15 ± 1.89 ψ | 2.92 ± 1.12 ψ π | |
| L-NAME-RF | 5.80 ± 0.83 | 4.49 ± 0.85 | 1.68 ± 0.38 ψ ϕ | |
| L-NAME-RF+NaHS | 5.64 ± 1.46 | 3.80 ± 0.78 | 1.63 ± 0.26 ψ | |
| L-NAME-RF+PAG | 5.58 ± 0.68 | 4.33 ± 0.62 | 1.58 ± 0.25 ψ ϕ |
| Groups | BW (gm) | KW (gm) | KI (%) |
|---|---|---|---|
| WKY-CONTROL | 317 ± 0.8 | 1.05 ± 0.05 | 0.3 ± 0.01 |
| WKY-RF | 282 ± 17.1 ψ | 1.04 ± 0.04 | 0.3 ± 0.02 ψ |
| WKY-RF+NaHS | 274 ± 6.9 | 0.94 ± 0.01 ϕ | 0.3 ± 0.00 |
| WKY-RF+PAG | 266 ± 5.3 | 1.01 ± 0.08 | 0.3 ± 0.02 |
| L-NAME-CONTROL | 284 ± 9.0 ψ | 1.08 ± 0.05 | 0.3 ± 0.01 |
| L-NAME-RF | 258 ± 6.8 ϕ # | 1.21 ± 0.02 ϕ # | 0.4 ± 0.02 ϕ # |
| L-NAME-RF+NaHS | 273 ± 3.4 | 1.13 ± 0.03 | 0.4± 0.01 |
| L-NAME-RF+PAG | 260 ± 8.1 | 1.19 ± 0.03 | 0.4 ± 0.01 π |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, A. Prophylactic Treatment with Hydrogen Sulphide Can Prevent Renal Ischemia-Reperfusion Injury in L-NAME Induced Hypertensive Rats with Cisplatin-Induced Acute Renal Failure. Life 2022, 12, 1819. https://doi.org/10.3390/life12111819
Ahmad A. Prophylactic Treatment with Hydrogen Sulphide Can Prevent Renal Ischemia-Reperfusion Injury in L-NAME Induced Hypertensive Rats with Cisplatin-Induced Acute Renal Failure. Life. 2022; 12(11):1819. https://doi.org/10.3390/life12111819
Chicago/Turabian StyleAhmad, Ashfaq. 2022. "Prophylactic Treatment with Hydrogen Sulphide Can Prevent Renal Ischemia-Reperfusion Injury in L-NAME Induced Hypertensive Rats with Cisplatin-Induced Acute Renal Failure" Life 12, no. 11: 1819. https://doi.org/10.3390/life12111819
APA StyleAhmad, A. (2022). Prophylactic Treatment with Hydrogen Sulphide Can Prevent Renal Ischemia-Reperfusion Injury in L-NAME Induced Hypertensive Rats with Cisplatin-Induced Acute Renal Failure. Life, 12(11), 1819. https://doi.org/10.3390/life12111819