
A Review Pertaining to SARS-CoV-2 and Autoimmune Diseases: What Is the Connection?


Abstract
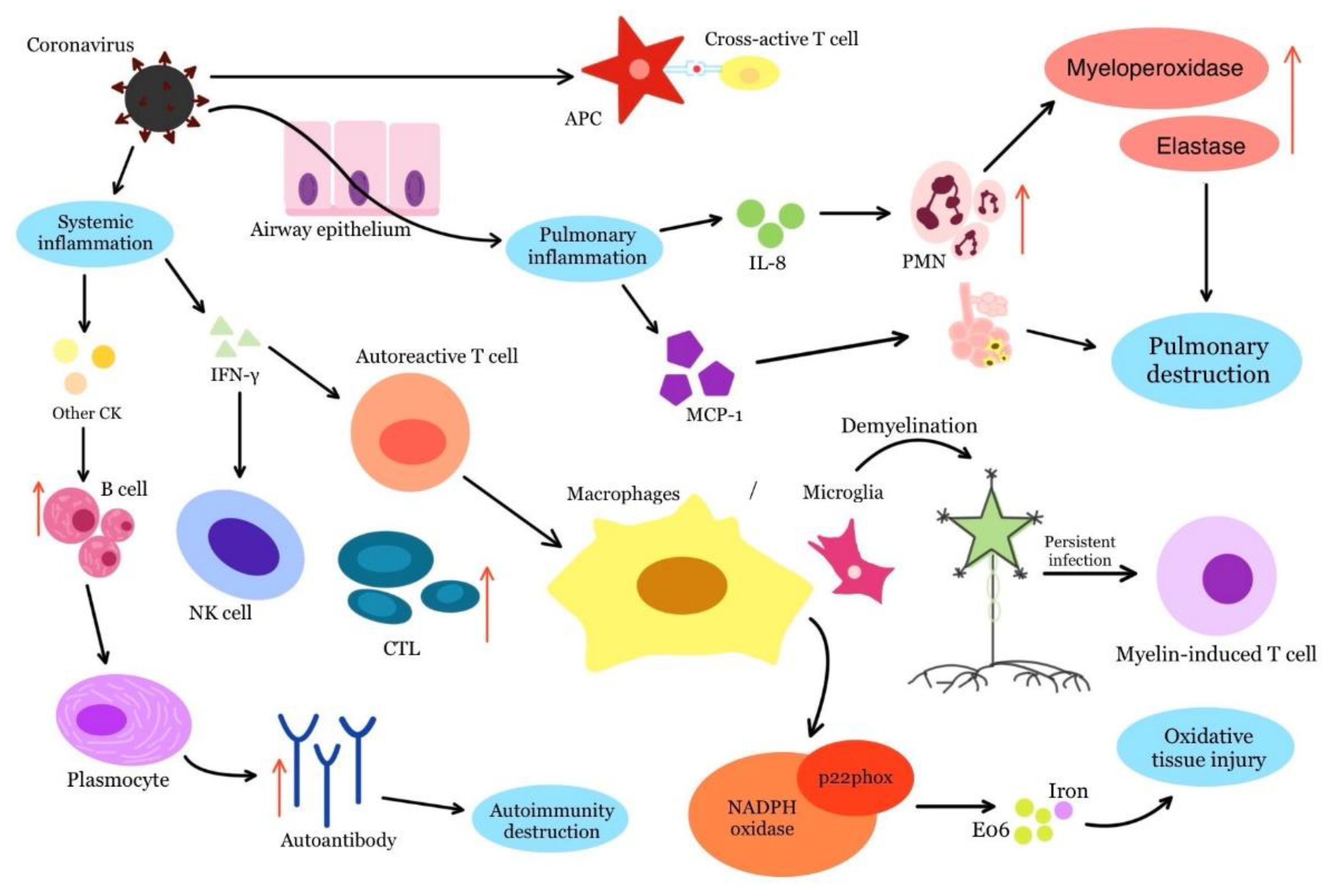
:1. Introduction
2. Connections between COVID-19 and Autoimmune Diseases
2.1. Systemic Lupus Erythematosus
2.2. Rheumatoid Arthritis
2.3. Multiple Sclerosis
3. Development of Autoimmune Diseases after SARS-CoV-2 Infection
3.1. Autoimmune Diseases after COVID-19
3.1.1. Guillain–Barré Syndrome
3.1.2. Antiphospholipid Syndrome
3.1.3. Kawasaki Disease in MIS-C
4. COVID-19 in Patients with Autoimmune Diseases
4.1. Systemic Lupus Erythematosus
4.2. Rheumatoid Arthritis
4.3. Multiple Sclerosis
4.4. Type 1 Diabetes Mellitus
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Saini, L.; Krishna, D.; Tiwari, S.; Goyal, J.P.; Kumar, P.; Khera, D.; Choudhary, B.; Didel, S.; Gadepalli, R.; Singh, K. Post-COVID-19 Immune-Mediated Neurological Complications in Children: An Ambispective Study. Pediatr. Neurol. 2022, 136, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.; Apgar, R.; Green, M. Cutaneous lupus-erythematosus-like reaction arising after COVID-19 vaccination. J. Cutan. Pathol. 2022, 49, 943–946. [Google Scholar] [CrossRef]
- Deora, H.; Dange, P.; Patel, K.; Shashidhar, A.; Tyagi, G.; Pruthi, N.; Arivazhagan, A.; Shukla, D.; Dwarakanath, S. Management of Neurosurgical Cases in a Tertiary Care Referral Hospital During the COVID-19 Pandemic: Lessons from a Middle-Income Country. World Neurosurg. 2021, 148, e197–e208. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H. Immunology of COVID-19. Environ. Microbiol. 2020, 22, 4895–4908. [Google Scholar] [CrossRef] [PubMed]
- Adil, M.T.; Rahman, R.; Whitelaw, D.; Jain, V.; Al-Taan, O.; Rashid, F.; Munasinghe, A.; Jambulingam, P. SARS-CoV-2 and the pandemic of COVID-19. Postgrad. Med. J. 2021, 97, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sawalha, A.H.; Lu, Q. COVID-19 and autoimmune diseases. Curr. Opin. Rheumatol. 2021, 33, 155–162. [Google Scholar] [CrossRef]
- Lakota, K.; Perdan-Pirkmajer, K.; Hočevar, A.; Sodin-Semrl, S.; Rotar, Ž.; Čučnik, S.; Žigon, P. COVID-19 in Association With Development, Course, and Treatment of Systemic Autoimmune Rheumatic Diseases. Front. Immunol. 2021, 11, 611318. [Google Scholar] [CrossRef]
- Velnar, T.; Bosnjak, R. Management of neurosurgical patients during coronavirus disease 2019 pandemics: The Ljubljana, Slovenia experience. World J. Clin. Cases 2022, 10, 4726–4736. [Google Scholar] [CrossRef]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Zhao, M.M.; Yang, W.L.; Yang, F.Y.; Zhang, L.; Huang, W.J.; Hou, W.; Fan, C.F.; Jin, R.H.; Feng, Y.M.; Wang, Y.C.; et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target. Ther. 2021, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, P.; Desikan, R.; Dixit, N.M. Targeting TMPRSS2 and Cathepsin B/L together may be synergistic against SARS-CoV-2 infection. PLoS Comput. Biol. 2020, 16, e1008461. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, S. How SARS-CoV-2 (COVID-19) spreads within infected hosts—What we know so far. Emerg. Top. Life Sci. 2020, 4, 371–378. [Google Scholar] [PubMed]
- Grange, L.; Guilpain, P.; Truchetet, M.-E.; Cracowski, J.-L. Challenges of autoimmune rheumatic disease treatment during the COVID-19 pandemic: A review. Therapies 2020, 75, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Pires, R.E.; Reis, I.G.N.; Waldolato, G.S.; Pires, D.D.; Bidolegui, F.; Giordano, V. What Do We Need to Know About Musculoskeletal Manifestations of COVID-19?: A Systematic Review. JBJS Rev. 2022, 10, e22.00013. [Google Scholar] [CrossRef]
- Russell, D.; Spence, N.J.; Chase, J.-A.D.; Schwartz, T.; Tumminello, C.M.; Bouldin, E. Support amid uncertainty: Long COVID illness experiences and the role of online communities. SSM-Qual. Res. Health 2022, 2, 100177. [Google Scholar] [CrossRef]
- Paterson, C.; Davis, D.; Roche, M.; Bissett, B.; Roberts, C.; Turner, M.; Baldock, E.; Mitchell, I. What are the long-term holistic health consequences of COVID-19 among survivors? An umbrella systematic review. J. Med. Virol. 2022, 94, 5653–5668. [Google Scholar] [CrossRef]
- Kraft, H.; Weiss, F. Pandemic portfolio choice. Eur. J. Oper. Res. 2023, 305, 451–462. [Google Scholar] [CrossRef]
- Capone, F.; Rossi, M.; Cruciani, A.; Motolese, F.; Pilato, F.; Di Lazzaro, V. Safety, immunogenicity, efficacy, and acceptability of COVID-19 vaccination in people with multiple sclerosis: A narrative review. Neural Regen. Res. 2023, 18, 284. [Google Scholar] [CrossRef]
- Lorenz, H.M.; Herrmann, M.; Kalden, J.R. The pathogenesis of autoimmune diseases. Scand. J. Clin. Lab. Investig. Suppl. 2001, 235, 16–26. [Google Scholar]
- Wang, L.; Wang, F.-S.; Gershwin, M.E. Human autoimmune diseases: A comprehensive update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef]
- Wu, X.; Wang, L.; Shen, L.; Tang, K. Response of COVID-19 vaccination in multiple sclerosis patients following disease-modifying therapies: A meta-analysis. eBioMedicine 2022, 81, 104102. [Google Scholar] [CrossRef] [PubMed]
- Lehuen, A.; Marshak-Rothstein, A. Organ-specific and systemic autoimmune diseases. Curr. Opin. Immunol. 2013, 25, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Seissler, J.; Scherbaum, W.A. Are we ready to predict and prevent endocrine/organ specific autoimmune diseases? Springer Semin. Immunopathol. 2002, 24, 273–295. [Google Scholar] [CrossRef]
- Dorward, D.A.; Russell, C.D.; Um, I.H.; Elshani, M.; Armstrong, S.D.; Penrice-Randal, R.; Millar, T.; Lerpiniere, C.E.B.; Tagliavini, G.; Hartley, C.S.; et al. Tissue-Specific Immunopathology in Fatal COVID-19. Am. J. Respir. Crit. Care Med. 2021, 203, 192–201. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; Gudjonsson, J.E.; Kahlenberg, J.M.; McCune, W.J.; Bockenstedt, P.L.; Karp, D.R.; Knight, J.S. Anti–Neutrophil Extracellular Trap Antibodies and Impaired Neutrophil Extracellular Trap Degradation in Antiphospholipid Syndrome. Arthritis Rheumatol. 2020, 72, 2130–2135. [Google Scholar] [CrossRef] [PubMed]
- Yalavarthi, S.; Gould, T.J.; Rao, A.N.; Mazza, L.F.; Morris, A.E.; Núñez-Álvarez, C.; Hernández-Ramírez, D.; Bockenstedt, P.L.; Liaw, P.C.; Cabral, A.R.; et al. Release of Neutrophil Extracellular Traps by Neutrophils Stimulated With Antiphospholipid Antibodies: A Newly Identified Mechanism of Thrombosis in the Antiphospholipid Syndrome. Arthritis Rheumatol. 2015, 67, 2990–3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagy, I.; Zeller, L.; Raviv, Y.; Porges, T.; Bieber, A.; Abu-Shakra, M. New-onset systemic lupus erythematosus following BNT162b2 mRNA COVID-19 vaccine: A case series and literature review. Rheumatol. Int. 2022, 42, 2261–2266. [Google Scholar] [CrossRef] [PubMed]
- Najafi, S.; Rajaei, E.; Moallemian, R.; Nokhostin, F. The potential similarities of COVID-19 and autoimmune disease pathogenesis and therapeutic options: New insights approach. Clin. Rheumatol. 2020, 39, 3223–3235. [Google Scholar] [CrossRef]
- Mok, C.C.; Lau, C.S. Pathogenesis of systemic lupus erythematosus. J. Clin. Pathol. 2003, 56, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Chen, L.; Lou, J.; Bai, Y.; Wang, M. COVID-19 Disease with Positive Fecal and Negative Pharyngeal and Sputum Viral Tests. Am. J. Gastroenterol. 2020, 115, 790. [Google Scholar] [CrossRef]
- Quintana, R.; Fernandez, S.; Guggia, L.; Fay, M.; Camacho, C.; Gomez, G.; Petrelli, J.; Honeri, A.; Solórzano, V.A.; Bensi, A.; et al. Social networks as education strategies for indigenous patients with rheumatoid arthritis during COVID-19 pandemic. Are they useful? Clin. Rheumatol. 2022, 41, 3313–3318. [Google Scholar] [CrossRef] [PubMed]
- Anić, B.; Mayer, M. Patogeneza reumatoidnog artritisa [Pathogenesis of rheumatoid arthritis]. Reumatizam 2014, 61, 19–23. [Google Scholar]
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir. Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- Di Filippo, M.; Gaetani, L. SARS-CoV-2 vaccines tolerability: A perspective by people with multiple sclerosis. Lancet Reg. Health Eur. 2022, 22, 100512. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Antel, J. Pathogenesis of multiple sclerosis. Curr. Opin. Neurol. 2005, 18, 225–230. [Google Scholar] [CrossRef]
- Miossec, P.; Kolls, J.K. Targeting IL-17 and TH17 cells in chronic inflammation. Nat. Rev. Drug Discov. 2012, 11, 763–776. [Google Scholar] [CrossRef]
- Maxeiner, H.-G.; Schneider, E.M.; Kurfiss, S.-T.; Brettschneider, J.; Tumani, H.; Bechter, K. Cerebrospinal fluid and serum cytokine profiling to detect immune control of infectious and inflammatory neurological and psychiatric diseases. Cytokine 2014, 69, 62–67. [Google Scholar] [CrossRef]
- do Prado, C.B.; Emerick, G.S.; Cevolani Pires, L.B.; Salaroli, L.B. Impact of long-term COVID on workers: A systematic review protocol. PLoS ONE 2022, 17, e0265705. [Google Scholar] [CrossRef] [PubMed]
- Castanares-Zapatero, D.; Chalon, P.; Kohn, L.; Dauvrin, M.; Detollenaere, J.; de Noordhout, C.M.; Jong, C.P.-D.; Cleemput, I.; Heede, K.V.D. Pathophysiology and mechanism of long COVID: A comprehensive review. Ann. Med. 2022, 54, 1473–1487. [Google Scholar] [CrossRef] [PubMed]
- Galeotti, C.; Bayry, J. Autoimmune and inflammatory diseases following COVID-19. Nat. Rev. Rheumatol. 2020, 16, 413–414. [Google Scholar] [CrossRef]
- Zhou, S.-Y.; Zhang, C.; Shu, W.-J.; Chong, L.-Y.; He, J.; Xu, Z.; Pan, H.-F. Emerging Roles of Coronavirus in Autoimmune Diseases. Arch. Med. Res. 2021, 52, 665–672. [Google Scholar] [CrossRef]
- Freire-de-Lima, L.; Scovino, A.M.; Barreto Menezes, C.C.; Marques da Fonseca, L.; Santos Dos Reis, J.; Rodrigues da Costa Santos, M.A.; Monteiro da Costa, K.; Antonio do Nascimento Santos, C.; Freire-de-Lima, C.G.; Morrot, A. Autoimmune Disorders & COVID-19. Medicines 2021, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.Y.; Mao, T.; Klein, J.; Dai, Y.; Huck, J.D.; Jaycox, J.R.; Liu, F.; Zhou, T.; Israelow, B.; Wong, P.; et al. Diverse functional autoantibodies in patients with COVID-19. Nature 2021, 595, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef] [PubMed]
- Dotan, A.; Muller, S.; Kanduc, D.; David, P.; Halpert, G.; Shoenfeld, Y. The SARS-CoV-2 as an instrumental trigger of autoimmunity. Autoimmun. Rev. 2021, 20, 102792. [Google Scholar] [CrossRef]
- Dalakas, M.C. Guillain-Barré syndrome: The first documented COVID-19-triggered autoimmune neurologic disease: More to come with myositis in the offing. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e781. [Google Scholar] [CrossRef]
- Sedaghat, Z.; Karimi, N. Guillain Barre syndrome associated with COVID-19 infection: A case report. J. Clin. Neurosci. 2020, 76, 233–235. [Google Scholar] [CrossRef]
- Wakerley, B.R.; Yuki, N. Infectious and noninfectious triggers in Guillain-Barre syndrome. Expert Rev. Clin. Immunol. 2013, 9, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Shen, D.; Zhou, H.; Liu, J.; Chen, S. Guillain-Barré syndrome associated with SARS-CoV-2 infection: Causality or coincidence? Lancet Neurol. 2020, 19, 383–384. [Google Scholar] [CrossRef]
- Serrano, M.; Espinosa, G.; Serrano, A.; Cervera, R. COVID-19 and the antiphospholipid syndrome. Autoimmun. Rev. 2022, 21, 103206. [Google Scholar] [CrossRef] [PubMed]
- Ehrenfeld, M.; Tincani, A.; Andreoli, L.; Cattalini, M.; Greenbaum, A.; Kanduc, D.; Alijotas-Reig, J.; Zinserling, V.; Semenova, N.; Amital, H.; et al. Covid-19 and autoimmunity. Autoimmun. Rev. 2020, 19, 102597. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, E.; Bramanti, A.; Ciurleo, R.; Tchorbanov, A.I.; Giordano, A.; Fagone, P.; Belizna, C.; Bramanti, P.; Shoenfeld, Y.; Nicoletti, F. Entangling COVID-19 associated thrombosis into a secondary antiphospholipid antibody syndrome: Diagnostic and therapeutic perspectives (Review). Int. J. Mol. Med. 2020, 46, 903–912. [Google Scholar] [CrossRef]
- Gkrouzman, E.; Barbhaiya, M.; Erkan, D.; Lockshin, M.D. Reality Check on Antiphospholipid Antibodies in COVID-19–Associated Coagulopathy. Arthritis Rheumatol. 2021, 73, 173–174. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Henry, B.M.; Lippi, G. COVID-19 and Antiphospholipid Antibodies: Time for a Reality Check? Semin. Thromb. Hemost. 2022, 48, 72–92. [Google Scholar] [CrossRef]
- Borghi, M.O.; Beltagy, A.; Garrafa, E.; Curreli, D.; Cecchini, G.; Bodio, C.; Grossi, C.; Blengino, S.; Tincani, A.; Franceschini, F.; et al. Anti-Phospholipid Antibodies in COVID-19 Are Different From Those Detectable in the Anti-Phospholipid Syndrome. Front. Immunol. 2020, 11, 584241. [Google Scholar] [CrossRef]
- Liu, J.; Li, F.; Shu, K.; Chen, T.; Wang, X.; Xie, Y.; Li, S.; Zhang, Z.; Jin, S.; Jiang, M. The analysis of false prolongation of the activated partial thromboplastin time (activator: Silica): Interference of C-reactive protein. J. Clin. Lab. Anal. 2018, 32, e22571. [Google Scholar] [CrossRef]
- Ramakers, C.; van der Heul, C.; van Wijk, E.M. Investigation of coagulation time: PT and APTT. Ned. Tijdschr. Geneeskd. 2012, 156, A3985. [Google Scholar]
- Pineton de Chambrun, M.; Frere, C.; Miyara, M.; Amoura, Z.; Martin-Toutain, I.; Mathian, A.; Hekimian, G.; Combes, A. High frequency of antiphospholipid antibodies in critically ill COVID-19 patients: A link with hypercoagulability? J. Intern. Med. 2021, 289, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Al-Beltagi, M.; Saeed, N.K.; Bediwy, A.S. COVID-19 disease and autoimmune disorders: A mutual pathway. World J. Methodol. 2022, 12, 200–223. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, C.; Hills, J.; Machin, S.J.; Cohen, H. Diagnosis of antiphospholipid syndrome in routine clinical practice. Lupus 2013, 22, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Bowles, L.; Platton, S.; Yartey, N.; Dave, M.; Lee, K.; Hart, D.P.; MacDonald, V.; Green, L.; Sivapalaratnam, S.; Pasi, K.J.; et al. Lupus Anticoagulant and Abnormal Coagulation Tests in Patients with COVID-19. N. Engl. J. Med. 2020, 383, 288–290. [Google Scholar] [CrossRef]
- Harzallah, I.; Debliquis, A.; Drénou, B. Lupus anticoagulant is frequent in patients with COVID-19. J. Thromb. Haemost. 2020, 18, 2064–2065. [Google Scholar] [CrossRef]
- Verdoni, L.; Mazza, A.; Gervasoni, A.; Martelli, L.; Ruggeri, M.; Ciuffreda, M.; Bonanomi, E.; D’Antiga, L. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: An observational cohort study. Lancet 2020, 395, 1771–1778. [Google Scholar] [CrossRef]
- De Ioris, M.A.; Scarselli, A.; Bracaglia, C.; Perrotta, D.; Bernardi, S.; Santilli, V.; Ceglie, G.; Fabozzi, F.; Agrati, C.; Prencipe, G.; et al. Common bone marrow signature in COVID-19-associated multisystem inflammatory syndrome in children: A first-wave small case series experience. Pediatr. Blood Cancer 2022, 69, e29919. [Google Scholar] [CrossRef]
- Riphagen, S.; Gomez, X.; Gonzalez-Martinez, C.; Wilkinson, N.; Theocharis, P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 2020, 395, 1607–1608. [Google Scholar] [CrossRef]
- Belhadjer, Z.; Méot, M.; Bajolle, F.; Khraiche, D.; Legendre, A.; Abakka, S.; Auriau, J.; Grimaud, M.; Oualha, M.; Beghetti, M.; et al. Acute Heart Failure in Multisystem Inflammatory Syndrome in Children in the Context of Global SARS-CoV-2 Pandemic. Circulation 2020, 142, 429–436. [Google Scholar] [CrossRef]
- McCrindle, B.W.; Rowley, A.H.; Newburger, J.W.; Burns, J.C.; Bolger, A.F.; Gewitz, M.; Baker, A.L.; Jackson, M.A.; Takahashi, M.; Shah, P.B.; et al. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals From the American Heart Association. Circulation 2017, 135, e927–e999. [Google Scholar] [CrossRef]
- Rodriguez-Gonzalez, M.; Castellano-Martinez, A.; Cascales-Poyatos, H.M.; Perez-Reviriego, A.A. Cardiovascular impact of COVID-19 with a focus on children: A systematic review. World J. Clin. Cases 2020, 8, 5250–5283. [Google Scholar] [CrossRef] [PubMed]
- Vukomanovic, V.; Krasic, S.; Minic, P.; Petrovic, G.; Nesic, D.; Paripovic, A.; Vasiljevic, M.; Gobeljic, B. Kawasaki-like disease and acute myocarditis in the SARS-CoV-2 pandemic—Reports of three adolescents. Bosn. J. Basic Med. Sci. 2021, 21, 252. [Google Scholar]
- Hromić-Jahjefendić, A.; Barh, D.; Ramalho Pinto, C.H.; Gabriel Rodrigues Gomes, L.; Picanço Machado, J.L.; Afolabi, O.O.; Tiwari, S.; Aljabali, A.A.A.; Tambuwala, M.M.; Serrano-Aroca, Á.; et al. Associations and Disease–Disease Interactions of COVID-19 with Congenital and Genetic Disorders: A Comprehensive Review. Viruses 2022, 14, 910. [Google Scholar] [CrossRef] [PubMed]
- Yongzhi, X. COVID-19-associated cytokine storm syndrome and diagnostic principles: An old and new Issue. Emerg. Microbes Infect. 2021, 10, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, H.; Li, J.; Liu, C.; Chen, H.; Li, J.; Sun, C.; Guo, T.; Pang, Z.; Zhang, B.; et al. Cytokine nanosponges suppressing overactive macrophages and dampening systematic cytokine storm for the treatment of hemophagocytic lymphohistiocytosis. Bioact. Mater. 2022, 21, 531–546. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Yang, S.L.; Xu, X.-J.; Tang, Y.-M.; Song, H.; Xu, W.-Q.; Zhao, F.-Y.; Shen, D.-Y. Associations between inflammatory cytokines and organ damage in pediatric patients with hemophagocytic lymphohistiocytosis. Cytokine 2016, 85, 14–17. [Google Scholar] [CrossRef]
- Zambrano, L.D.; Ly, K.N.; Link-Gelles, R.; Newhams, M.M.; Akande, M.; Wu, M.J.; Feldstein, L.R.; Tarquinio, K.M.; Sahni, L.C.; Riggs, B.J.; et al. Overcoming COVID-19 Investigators. Investigating Health Disparities Associated With Multisystem Inflammatory Syndrome in Children After SARS-CoV-2 Infection. Pediatr. Infect. Dis. J. 2022, 41, 891–898. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Paludan, S.R.; Mogensen, T.H. Innate immunological pathways in COVID-19 pathogenesis. Sci. Immunol. 2022, 7, 5505. [Google Scholar] [CrossRef]
- Li, H.; Liu, L.; Zhang, D.; Xu, J.; Dai, H.; Tang, N.; Su, X.; Cao, B. SARS-CoV-2 and viral sepsis: Observations and hypotheses. Lancet 2020, 395, 1517–1520. [Google Scholar] [CrossRef]
- Bastard, P.; Michailidis, E.; Hoffmann, H.H.; Chbihi, M.; Le Voyer, T.; Rosain, J.; Philippot, Q.; Seeleuthner, Y.; Gervais, A.; Materna, M.; et al. Auto-antibodies to type I IFNs can underlie adverse reactions to yellow fever live attenuated vaccine. J. Exp. Med. 2021, 218, e20202486. [Google Scholar] [CrossRef] [PubMed]
- Carter-Timofte, M.E.; Jorgensen, S.E.; Freytag, M.R.; Thomsen, M.M.; Brinck Andersen, N.S.; Al-Mousawi, A.; Hait, A.S.; Mogensen, T.H. Deciphering the role of host genetics in susceptibility to severe COVID-19. Front. Immunol. 2020, 11, 1606. [Google Scholar] [CrossRef] [PubMed]
- Moreews, M.; Le Gouge, K.; Khaldi-Plassart, S.; Pescarmona, R.; Mathieu, A.L.; Malcus, C.; Djebali, S.; Bellomo, A.; Dauwalder, O.; Perret, M.; et al. Polyclonal expansion of TCR Vbeta 21.3(+) CD4(+) and CD8(+) T cells is a hallmark of multisystem inflammatory syndrome in children. Sci. Immunol. 2021, 6, eabh1516. [Google Scholar] [CrossRef] [PubMed]
- Schnaubelt, S.; Tihanyi, D.; Strassl, R.; Schmidt, R.; Anders, S.; Laggner, A.N.; Agis, H.; Domanovits, H. Hemophagocytic Lymphohistiocytosis in COVID-19: Case Reports of a Stepwise Approach. Medicine 2021, 100, e25170. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider Cytokine Storm Syndromes and Immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Ihara, B.P.; Lindoso, L.M.; Setoue, D.N.D.; Tanigava, N.Y.; Helito, A.C.; Simon, J.R.; Viana, V.S.L.; Strabelli, C.A.A.; Pedroso, C.A.A.; Sieczkowska, S.M.; et al. Campos LMMA. COVID-19 quarantine in adolescents with autoimmune rheumatic diseases: Mental health issues and life conditions. Clin. Rheumatol. 2022, 41, 3189–3198. [Google Scholar] [CrossRef]
- Babtain, F.; Bajafar, A.; Nazmi, O.; Badawi, M.; Basndwah, A.; Bushnag, A.; Cupler, E.; Hassan, A. The disease course of multiple sclerosis before and during COVID-19 pandemic: A retrospective five-year study. Mult. Scler. Relat. Disord. 2022, 65, 103985. [Google Scholar] [CrossRef]
- Montero, F.; Martínez-Barrio, J.; Serrano-Benavente, B.; González, T.; Rivera, J.; Molina Collada, J.; Castrejón, I.; Álvaro-Gracia, J. Coronavirus disease 2019 (COVID-19) in autoimmune and inflammatory conditions: Clinical characteristics of poor outcomes. Rheumatol. Int. 2020, 40, 1593–1598. [Google Scholar] [CrossRef]
- Barcellini, W.; Giannotta, J.A.; Fattizzo, B. Are Patients With Autoimmune Cytopenias at Higher Risk of COVID-19 Pneumonia? The Experience of a Reference Center in Northern Italy and Review of the Literature. Front. Immunol. 2021, 11, 609198. [Google Scholar] [CrossRef]
- Akiyama, S.; Hamdeh, S.; Micic, D.; Sakuraba, A. Prevalence and clinical outcomes of COVID-19 in patients with autoimmune diseases: A systematic review and meta-analysis. Ann. Rheum. Dis. 2021, 80, 384–391. [Google Scholar] [CrossRef]
- Bakasis, A.-D.; Mavragani, C.P.; Boki, K.A.; Tzioufas, A.G.; Vlachoyiannopoulos, P.G.; Stergiou, I.E.; Skopouli, F.N.; Moutsopoulos, H.M. COVID-19 infection among autoimmune rheumatic disease patients: Data from an observational study and literature review. J. Autoimmun. 2021, 123, 102687. [Google Scholar] [CrossRef] [PubMed]
- Voiriot, G.; Philippot, Q.; Elabbadi, A.; Elbim, C.; Chalumeau, M.; Fartoukh, M. Risks Related to the Use of Non-Steroidal Anti-Inflammatory Drugs in Community-Acquired Pneumonia in Adult and Pediatric Patients. J. Clin. Med. 2019, 8, 786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Chen, C.; Hu, F.; Wang, J.; Zhao, Q.; Gale, R.P.; Liang, Y. Impact of corticosteroid therapy on outcomes of persons with SARS-CoV-2, SARS-CoV, or MERS-CoV infection: A systematic review and meta-analysis. Leukemia 2020, 34, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; Ahmed, M.; Conway, R.; Carey, J.J. Risk of Infection with Methotrexate Therapy in Inflammatory Diseases: A Systematic Review and Meta-Analysis. J. Clin. Med. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathian, A.; Mahevas, M.; Rohmer, J.; Roumier, M.; Cohen-Aubart, F.; Amador-Borrero, B.; Barrelet, A.; Chauvet, C.; Chazal, T.; Delahousse, M.; et al. Clinical course of coronavirus disease 2019 (COVID-19) in a series of 17 patients with systemic lupus erythematosus under long-term treatment with hydroxychloroquine. Ann. Rheum. Dis. 2020, 79, 837–839. [Google Scholar] [CrossRef]
- Boulware, D.R.; Pullen, M.F.; Bangdiwala, A.S.; Pastick, K.A.; Lofgren, S.M.; Okafor, E.C.; Skipper, C.P.; Nascene, A.A.; Nicol, M.R.; Abassi, M.; et al. A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for Covid-19. N. Engl. J. Med. 2020, 383, 517–525. [Google Scholar] [CrossRef]
- Wallace, B.; Washer, L.; Marder, W.; Kahlenberg, J.M. Patients with lupus with COVID-19: University of Michigan experience. Ann. Rheum. Dis. 2020, 80, e35. [Google Scholar] [CrossRef]
- Konig, M.F.; Kim, A.H.; Scheetz, M.H.; Graef, E.R.; Liew, J.W.; Simard, J.; Machado, P.M.; Gianfrancesco, M.; Yazdany, J.; Langguth, D.; et al. COVID-19 Global Rheumatology Alliance. Baseline use of hydroxychloroquine in systemic lupus erythematosus does not preclude SARS-CoV-2 infection and severe COVID-19. Ann. Rheum. Dis. 2020, 79, 1386–1388. [Google Scholar] [CrossRef]
- Gianfrancesco, M.; Hyrich, K.L.; Al-Adely, S.; Carmona, L.; Danila, M.I.; Gossec, L.; Izadi, Z.; Jacobsohn, L.; Katz, P.; Lawson-Tovey, S.; et al. Characteristics associated with hospitalisation for COVID-19 in people with rheumatic disease: Data from the COVID-19 Global Rheumatology Alliance physician-reported registry. Ann. Rheum. Dis. 2020, 79, 859–866. [Google Scholar] [CrossRef]
- Gautret, P.; Hoang, V.T.; Lagier, J.-C.; Raoult, D. Effect of hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial, an update with an intention-to-treat analysis and clinical outcomes. Int. J. Antimicrob. Agents 2021, 57, 106239. [Google Scholar] [CrossRef]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of Angiotensin-Converting Enzyme Inhibition and Angiotensin II Receptor Blockers on Cardiac Angiotensin-Converting Enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [Green Version]
- Biot, C.; Daher, W.; Chavain, N.; Fandeur, T.; Khalife, J.; Dive, D.; De Clercq, E. Design and Synthesis of Hydroxyferroquine Derivatives with Antimalarial and Antiviral Activities. J. Med. Chem. 2006, 49, 2845–2849. [Google Scholar] [CrossRef] [PubMed]
- Taplin, C.E.; Barker, J.M. Autoantibodies in type 1 diabetes. Autoimmunity 2008, 41, 11–18. [Google Scholar] [CrossRef]
- Paschou, S.A.; Papadopoulou-Marketou, N.; Chrousos, G.P.; Kanaka-Gantenbein, C. On type 1 diabetes mellitus pathogenesis. Endocr. Connect. 2018, 7, R38–R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, S.-Q.; Peng, H.-J. Characteristics of and Public Health Responses to the Coronavirus Disease 2019 Outbreak in China. J. Clin. Med. 2020, 9, 575. [Google Scholar] [CrossRef]


{kind=link}
| Autoantibody | Clinical Significance |
|---|---|
| Anti-IFN-I antibody | Causes severe forms of COVID-19 |
| Anti-IFN-α antibody | Possible emergence of autoimmune diseases and worsening of active COVID-19 infection |
| ANA | Poor prognosis |
| aPL | Poor prognosis |
| Anti-A2Ab | Worsens pulmonary symptoms during COVID-19 infection |
| Classic KD | KD-COVID-19 | |
|---|---|---|
| Age | patients younger than 5 years | older patients (~7.5 years) |
| Gastrointestinal and meningeal manifestations | atypical | usually present |
| Blood count | leukocytosis, mild normocytic anemia, thrombocytosis, elevated erythrocyte sedimentation rate or CRP | leukopenia with marked lymphopenia, thrombocytopenia |
| Procalcitonin, ferritin, cardiac enzymes, troponin | elevated | even higher |
| Signs of myocarditis | may occur | increased incidence |
| Coronary angiograms | coronary artery aneurysms may lead to myocardial ischemia | larger aneurysmal dilatations of coronary arteries with resultant thrombosis and myocardial infarction |
| Steroid therapy | may be used | is commonly used |
| Effect of IVIG on CAAs | good response | resistance to therapy is common |
| Autoimmune Diseases | Medications | Drug Function | Response in Autoimmune Patients | Possible Response in COVID-19 Patients |
|---|---|---|---|---|
| Systemic lupus erythematosus | Fedratinib | JAK2 inhibitor | Good | Good (could possibly reduce the risk of cytokine storm) |
| Anifrolumab | Antibody against the IFN-α receptor | Good | Good (could possibly reduce the risk of cytokine storm) | |
| Chloroquine and hydroxychloroquine | Antimalarial drugs that have negative effect on viral binding to the receptor and its replication | Good | Good (interferes with ACE2 glycolysis and reduces cytokine storm formation) | |
| Belimumab | Antibody against soluble stimulator of lymphocyte B | Good | Good (reduces the intensity of the immune response) | |
| Rheumatoid arthritis | Tocilizumab and sarilumab | Antibodies against the IL-6 receptor | Good | Good (inhibits the IL-6 binding to its receptors and reduces the cytokines inflammatory activity) |
| Quinapril and ramipril | ACE inhibitors | Good | Good (prevents the angiotensin II production and its associated inflammatory effects) | |
| Multiple sclerosis | Alemtuzumab | Anti-CD52 | Good | Not good (removes T cells, which help prevent viral infections) |
| Ocrelizumab and rituximab | Anti-CD20 | Good | Good (reduces the amount of interleukins) | |
| Tocilizumab | IL-6 receptor blocker | Good | Good (could possibly reduce the risk of cytokine storm) | |
| Etanercept | Anti-TNFα | Not good | Good (could possibly reduce the risk of cytokine storm) | |
| Natalizumab | Antibody against CD49d | Good | Good (limits leukocyte binding and trafficking) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kocivnik, N.; Velnar, T. A Review Pertaining to SARS-CoV-2 and Autoimmune Diseases: What Is the Connection? Life 2022, 12, 1918. https://doi.org/10.3390/life12111918
Kocivnik N, Velnar T. A Review Pertaining to SARS-CoV-2 and Autoimmune Diseases: What Is the Connection? Life. 2022; 12(11):1918. https://doi.org/10.3390/life12111918
Chicago/Turabian StyleKocivnik, Nina, and Tomaz Velnar. 2022. "A Review Pertaining to SARS-CoV-2 and Autoimmune Diseases: What Is the Connection?" Life 12, no. 11: 1918. https://doi.org/10.3390/life12111918
APA StyleKocivnik, N., & Velnar, T. (2022). A Review Pertaining to SARS-CoV-2 and Autoimmune Diseases: What Is the Connection? Life, 12(11), 1918. https://doi.org/10.3390/life12111918