
From Samples to Germline and Somatic Sequence Variation: A Focus on Next-Generation Sequencing in Melanoma Research

 , , , , , , and
, , , , , , and 
Abstract
:1. Introduction
2. DNA Libraries
3. RNA Libraries
4. Sequencing-Based Approaches in Cancer and Cutaneous Melanoma Research
4.1. Sequencing with the Classic Approaches
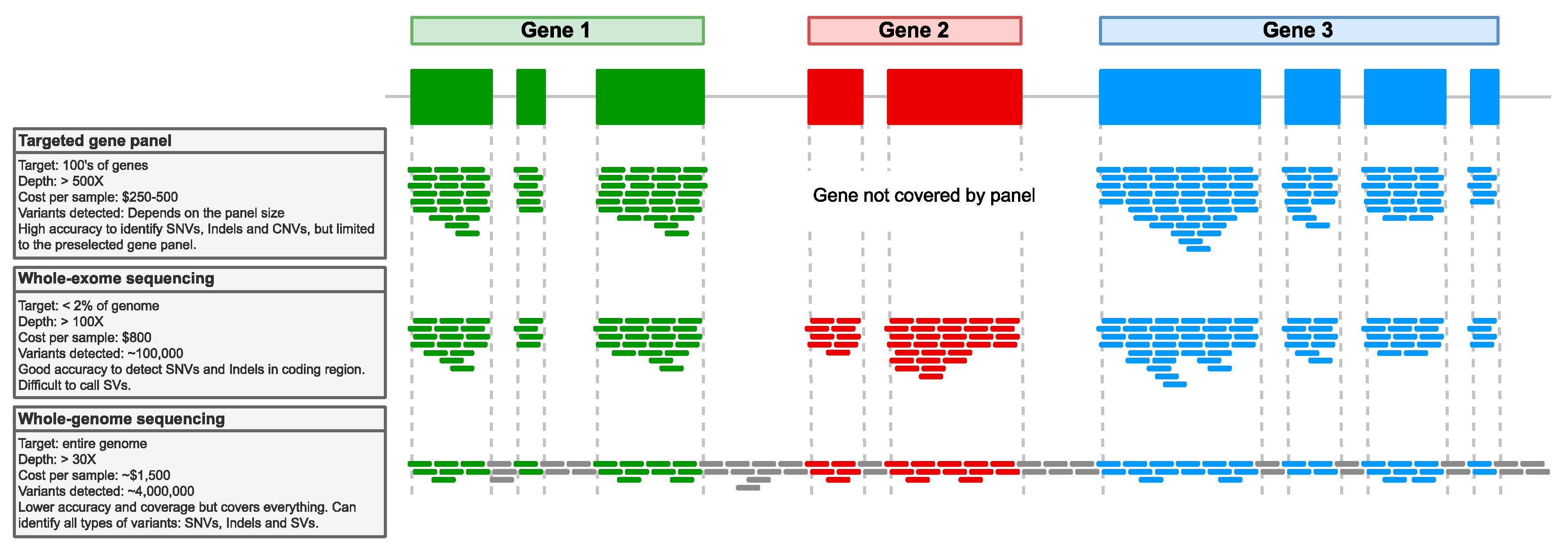
4.2. Next-Generation Sequencing
5. Bioinformatic Workflows for NGS Data Analysis
5.1. Read Alignment to the Reference Genome
5.2. Variant Calling of SNVs and Indels

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Somatic Callers | Sequencing Approach | Type Mutations | Normal Sample Required in Somatic Mode | Related Somatic Studies | ||
|---|---|---|---|---|---|---|
| Targeted | WES | WGS | ||||
| GATK-Mutect2 [105] | ✓ | ✓ | ✓ | SNVs and indels | Optional | Liver cancer [128], lung cancer [129] |
| Strelka2 [125] | ✓ | ✓ | ✓ | SNVs and indels | Yes | Cervical cancer [130] |
| VarDict [131] | ✓ | ✓ | ✓ | SNVs and indels | Optional | Breast and ovarian cancer [132] |
| CNVKit [133] | ✓ | ✓ | ✓ | CNVs | No | Melanoma [134] |
| Manta [135] | ✓ | ✓ | ✓ | SNVs and indels | Optional | Gastric cancer [136] |
| Delly [137] | x | x | ✓ | SVs | Yes | Plantar melanoma [138] |
| Lumpy [139] | x | x | ✓ | SVs | Optional | Colon cancer [140] |
| GRIDSS [141] | ✓ | ✓ | ✓ | SVs | Yes | Myeloid leukemia [142] |
| Varscan2 [126] | x | ✓ | x | SNVs and indels | Yes | Uveal melanoma [143] |
| ClinCNV [144] | ✓ | ✓ | ✓ | CNVs | Yes | Cutaneous leukemia [145] |
| ExomeDepth [146] | ✓ | ✓ | x | CNVs | No | Breast cancer [147] |
| ClinSV [148] | x | x | ✓ | SVs | No | Breast cancer [149] |
5.3. Variant Calling of SVs and CNVs
5.4. Variant Annotation, Filtering, and Prioritization
5.5. Tumor Clone Identification
5.6. Gene Fusions
5.7. Further Quality Control Steps to Perform in the Callset
5.7.1. Relatedness
5.7.2. Sex Inference
6. Long-Read Sequencing Technologies in Cancer Genomics
6.1. Advantages and Limitations of Long-Read Sequencing in Cancer Genomics
6.2. An exemplar Application of WGS with Long Reads from ONT
6.2.1. Library Preparation and Sequencing
6.2.2. Bioinformatic Tools for Long-Read Analysis
6.2.3. De Novo Genome Assembly
6.2.4. SV Calling
7. Discussion
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ottaviano, M.; Giunta, E.; Tortora, M.; Curvietto, M.; Attademo, L.; Bosso, D.; Cardalesi, C.; Rosanova, M.; De Placido, P.; Pietroluongo, E.; et al. BRAF Gene and Melanoma: Back to the Future. Int. J. Mol. Sci. 2021, 22, 3474. [Google Scholar] [CrossRef] [PubMed]
- Walia, V.; Mu, E.W.; Lin, J.C.; Samuels, Y. Delving into somatic variation in sporadic melanoma. Pigment Cell Melanoma Res. 2012, 25, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Peris, K.; Hauschild, A.; Saiag, P.; Middleton, M.; Bastholt, L.; Grob, J.-J.; Malvehy, J.; Newton-Bishop, J.; Stratigos, A.J.; et al. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline—Update 2016. Eur. J. Cancer 2016, 63, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Gandini, S.; Sera, F.; Cattaruzza, M.S.; Pasquini, P.; Abeni, D.; Boyle, P.; Melchi, C.F. Meta-analysis of risk factors for cutaneous melanoma: I. Common and atypical naevi. Eur. J. Cancer 2005, 41, 28–44. [Google Scholar] [CrossRef] [Green Version]
- Bataille, V.; Bishop, J.A.; Sasieni, P.; Swerdlow, A.; Pinney, E.; Griffiths, K.M.; Cuzick, J. Risk of cutaneous melanoma in relation to the numbers, types and sites of naevi: A case-control study. Br. J. Cancer 1996, 73, 1605–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krengel, S.; Hauschild, A.; Schäfer, T. Melanoma risk in congenital melanocytic naevi: A systematic review. Br. J. Dermatol. 2006, 155, 1–8. [Google Scholar] [CrossRef]
- Gandini, S.; Sera, F.; Cattaruzza, M.S.; Pasquini, P.; Picconi, O.; Boyle, P.; Melchi, C.F. Meta-analysis of risk factors for cutaneous melanoma: II. Sun exposure. Eur. J. Cancer 2005, 41, 45–60. [Google Scholar] [CrossRef]
- Greinert, R. Skin Cancer: New Markers for Better Prevention. Pathobiology 2009, 76, 64–81. [Google Scholar] [CrossRef]
- Jemal, A.; Devesa, S.S.; Fears, T.R.; Hartge, P. Cancer surveillance series: Changing patterns of cutaneous malignant melanoma mortality rates among whites in the United States. JNCI J. Natl. Cancer Inst. 2000, 92, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Tucker, M.A. Is Sunlight Important to Melanoma Causation? Cancer Epidemiol. Biomark. Prev. 2008, 17, 467–468. [Google Scholar] [CrossRef]
- Mitra, D.; Luo, X.; Morgan, A.; Wang, J.; Hoang, M.P.; Lo, J.; Guerrero, C.R.; Lennerz, J.K.; Mihm, M.C.; Wargo, J.A.; et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 2012, 491, 449–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, S.; Sera, F.; Gandini, S.; Iodice, S.; Caini, S.; Maisonneuve, P.; Fargnoli, M.C. MC1R variants, melanoma and red hair color phenotype: A meta-analysis. Int. J. Cancer 2008, 122, 2753–2760. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakob, J.A.; Bassett, R.L.; Ng, C.S.; Curry, J.L.; Joseph, R.; Alvarado, G.C.; Apn, M.L.R.; Richard, J.; Gershenwald, J.E.; Kim, K.B.; et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 2011, 118, 4014–4023. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic Activation of KIT in Distinct Subtypes of Melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef]
- Krauthammer, M.; Kong, Y.; Bacchiocchi, A.; Evans, P.; Pornputtapong, N.; Wu, C.; McCusker, J.P.; Ma, S.; Cheng, E.; Straub, R.; et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet. 2015, 47, 996–1002. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Pellegrini, C.; Cardelli, L.; Ciciarelli, V.; di Nardo, L.; Fargnoli, M.C. Familial melanoma: Diagnostic and management implications. Dermatol. Pract. Concept. 2019, 9, 10–16. [Google Scholar] [CrossRef]
- Rusch, M.; Nakitandwe, J.; Shurtleff, S.; Newman, S.; Zhang, Z.; Edmonson, M.N.; Parker, M.; Jiao, Y.; Ma, X.; Liu, Y.; et al. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Nattestad, M.; Goodwin, S.; Ng, K.; Baslan, T.; Sedlazeck, F.J.; Rescheneder, P.; Garvin, T.; Fang, H.; Gurtowski, J.; Hutton, E.; et al. Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line. Genome Res. 2018, 28, 1126–1135. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; Suzuki, M.; Mizushima-Sugano, J.; Frith, M.; Makałowski, W.; Kohno, T.; Sugano, S.; Tsuchihara, K.; Suzuki, Y. Sequencing and phasing cancer mutations in lung cancers using a long-read portable sequencer. DNA Res. 2017, 24, 585–596. [Google Scholar] [CrossRef]
- Vanni, I.; Tanda, E.T.; Spagnolo, F.; Andreotti, V.; Bruno, W.; Ghiorzo, P. The Current State of Molecular Testing in the BRAF-Mutated Melanoma Landscape. Front. Mol. Biosci. 2020, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Lightbody, G.; Haberland, V.; Browne, F.; Taggart, L.; Zheng, H.; Parkes, E.; Blayney, J.K. Review of applications of high-throughput sequencing in personalized medicine: Barriers and facilitators of future progress in research and clinical application. Brief. Bioinform. 2019, 20, 1795–1811. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, Y.; Nakamura, T.; Satonaka, K.; Maeda, S.; Doi, K.; Baba, S.; Sugiyama, T. Fundamental study on the mechanism of DNA degradation in tissues fixed in formaldehyde. J. Clin. Pathol. 1990, 43, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Do, H.; Dobrovic, A. Sequence Artifacts in DNA from Formalin-Fixed Tissues: Causes and Strategies for Minimization. Clin. Chem. 2015, 61, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Eckhart, L.; Bach, J.; Ban, J.; Tschachler, E. Melanin Binds Reversibly to Thermostable DNA Polymerase and Inhibits Its Activity. Biochem. Biophys. Res. Commun. 2000, 271, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Guyard, A.; Boyez, A.; Pujals, A.; Robe, C.; Van Nhieu, J.T.; Allory, Y.; Moroch, J.; Georges, O.; Fournet, J.-C.; Zafrani, E.-S.; et al. DNA degrades during storage in formalin-fixed and paraffin-embedded tissue blocks. Virchows Arch. 2017, 471, 491–500. [Google Scholar] [CrossRef]
- Ludyga, N.; Grünwald, B.; Azimzadeh, O.; Englert, S.; Höfler, H.; Tapio, S.; Aubele, M. Nucleic acids from long-term preserved FFPE tissues are suitable for downstream analyses. Virchows Arch. 2012, 460, 131–140. [Google Scholar] [CrossRef]
- Millán-Esteban, D.; Reyes-García, D.; García-Casado, Z.; Bañuls, J.; López-Guerrero, J.A.; Requena, C.; Rodríguez-Hernández, A.; Traves, V.; Nagore, E. Suitability of melanoma FFPE samples for NGS libraries: Time and quality thresholds for downstream molecular tests. BioTechniques 2018, 65, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Mathieson, W.; Thomas, G.A. Why Formalin-fixed, Paraffin-embedded Biospecimens Must Be Used in Genomic Medicine: An Evidence-based Review and Conclusion. J. Histochem. Cytochem. 2020, 68, 543–552. [Google Scholar] [CrossRef]
- van Dijk, E.L.; Jaszczyszyn, Y.; Thermes, C. Library preparation methods for next-generation sequencing: Tone down the bias. Exp. Cell Res. 2014, 322, 12–20. [Google Scholar] [CrossRef]
- Petty, D.R.; Hassan, O.A.; Barker, C.S.; O’Neill, S.S. Rapid BRAF Mutation Testing in Pigmented Melanomas. Am. J. Dermatopathol. 2019, 42, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Frouin, E.; Maudelonde, T.; Senal, R.; Larrieux, M.; Costes, V.; Godreuil, S.; Vendrell, J.A.; Solassol, J. Comparative Methods to Improve the Detection of BRAF V600 Mutations in Highly Pigmented Melanoma Specimens. PLoS ONE 2016, 11, e0158698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente, A.L.S.A.; Bianchini, R.A.; Laus, A.C.; Macedo, G.; Reis, R.M.; de Lima Vazquez, V. Comparison of protocols for removal of melanin from genomic DNA to optimize PCR amplification of DNA purified from highly pigmented lesions. Histol. Histopathol. Cell Biol. Tissue Eng. 2019, 34, 1089–1096. [Google Scholar] [CrossRef]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 2015, 951–969. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Zhou, Q.; Chen, Y. Impact of RNA degradation on next-generation sequencing transcriptome data. Genomics 2022, 114, 110429. [Google Scholar] [CrossRef]
- Rudloff, U.; Bhanot, U.; Gerald, W.; Klimstra, D.S.; Jarnagin, W.R.; Brennan, M.; Allen, P.J. Biobanking of Human Pancreas Cancer Tissue: Impact of Ex-Vivo Procurement Times on RNA Quality. Ann. Surg. Oncol. 2010, 17, 2229–2236. [Google Scholar] [CrossRef] [Green Version]
- Völler, D.; Reinders, J.; Meister, G.; Bosserhoff, A.-K. Strong reduction of AGO2 expression in melanoma and cellular consequences. Br. J. Cancer 2013, 109, 3116–3124. [Google Scholar] [CrossRef] [Green Version]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [Green Version]
- Maria, M.; Ajmal, M.; Azam, M.; Waheed, N.K.; Siddiqui, S.N.; Mustafa, B.; Ayub, H.; Ali, L.; Ahmad, S.; Micheal, S.; et al. Homozygosity Mapping and Targeted Sanger Sequencing Reveal Genetic Defects Underlying Inherited Retinal Disease in Families from Pakistan. PLoS ONE 2015, 10, e0119806. [Google Scholar] [CrossRef] [Green Version]
- Bezdíčka, M.; Štolbová; Seeman, T.; Cinek, O.; Malina, M.; Šimánková, N.; Průhová, .; Zieg, J. Genetic diagnosis of steroid-resistant nephrotic syndrome in a longitudinal collection of Czech and Slovak patients: A high proportion of causative variants in NUP93. Pediatr. Nephrol. 2018, 33, 1347–1363. [Google Scholar] [CrossRef]
- Liang, C.; Wu, Z.; Gan, X.; Liu, Y.; You, Y.; Liu, C.; Zhou, C.; Liang, Y.; Mo, H.; Chen, A.M.; et al. Detection of Rare Mutations in EGFR-ARMS-PCR-Negative Lung Adenocarcinoma by Sanger Sequencing. Yonsei Med. J. 2018, 59, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Bruijns, B.; Tiggelaar, R.M.; Gardeniers, J. Massively parallel sequencing techniques for forensics: A review. Electrophoresis 2018, 39, 2642–2654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Human Genome Sequencing Consortium. International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef] [Green Version]
- Sheen, Y.-S.; Liao, Y.-H.; Liau, J.-Y.; Lin, M.-H.; Hsieh, Y.-C.; Jee, S.-H.; Chu, C.-Y. Prevalence of BRAF and NRAS mutations in cutaneous melanoma patients in Taiwan. J. Formos. Med. Assoc. 2016, 115, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Ren, M.; Zhang, J.; Kong, Y.; Bai, Q.; Qi, P.; Wang, Q.; Zhou, X.; Chen, Y.; Zhu, X. BRAF, C-KIT, and NRAS mutations correlated with different clinicopathological features: An analysis of 691 melanoma patients from a single center. Ann. Transl. Med. 2022, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Fatnassi-Mersni, G.; Arfaoui, A.T.; Cherni, M.; Jones, M.; Zeglaoui, F.; Ouzari, H.I.; Rammeh, S. Molecular and immunohistochemical analysis of BRAF gene in primary cutaneous melanoma: Discovery of novel mutations. J. Cutan. Pathol. 2020, 47, 794–799. [Google Scholar] [CrossRef]
- Cheng, L.Y.; Haydu, L.E.; Song, P.; Nie, J.; Tetzlaff, M.T.; Kwong, L.N.; Gershenwald, J.E.; Davies, M.A.; Zhang, D.Y. High sensitivity sanger sequencing detection of BRAF mutations in metastatic melanoma FFPE tissue specimens. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Chen, C.; Fangxuqian, X.; Sun, S. Diagnosis of polyglutamine spinocerebellar ataxias by polymerase chain reaction amplification and Sanger sequencing. Mol. Med. Rep. 2018, 18, 1037–1042. [Google Scholar] [CrossRef]
- Fukuta, M.; Gaballah, M.; Takada, K.; Miyazaki, H.; Kato, H.; Aoki, Y.; Hamed, S.S.; ElMorsi, D.A.; ElDakroory, S.A. Genetic polymorphism of 27 X-chromosomal short tandem repeats in an Egyptian population. Leg. Med. 2019, 37, 64–66. [Google Scholar] [CrossRef]
- Khan, A.A.; Perveen, R.; Sheikh, N.; Abbasi, B.H.A.; Batool, Z.; Shahzad, M.; Kaleem, S. Genetic polymorphism of 15 autosomal short tandem repeats in Baloch population of Pakistan. Int. J. Legal Med. 2018, 133, 775–776. [Google Scholar] [CrossRef]
- Nyren, P.; Pettersson, B.; Uhlen, M. Solid Phase DNA Minisequencing by an Enzymatic Luminometric Inorganic Pyrophosphate Detection Assay. Anal. Biochem. 1993, 208, 171–175. [Google Scholar] [CrossRef]
- Edlundhrose, E.; Egyhazi, S.; Omholt, K.; Manssonbrahme, E.; Platz, A.; Hansson, J.; Lundeberg, J. NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: A study based on mutation screening by pyrosequencing. Melanoma Res. 2006, 16, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Yaman, B.; Kandiloğlu, G.; Akalin, T. BRAF-V600 Mutation Heterogeneity in Primary and Metastatic Melanoma. Am. J. Dermatopathol. 2016, 38, 113–120. [Google Scholar] [CrossRef]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Cohen, J.S.; Vernon, H.; Barañano, K.; McClellan, R.; Jamal, L.; Naidu, S.; Fatemi, A. Clinical whole exome sequencing in child neurology practice. Ann. Neurol. 2014, 76, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Muzny, D.M.; Xia, F.; Niu, Z.; Person, R.; Ding, Y.; Ward, P.; Braxton, A.; Wang, M.; Buhay, C.; et al. Molecular Findings Among Patients Referred for Clinical Whole-Exome Sequencing. JAMA 2014, 312, 1870–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vissers, L.E.; van Nimwegen, K.J.; Schieving, J.H.; Kamsteeg, E.-J.; Kleefstra, T.; Yntema, H.G.; Pfundt, R.; van der Wilt, G.J.; Krabbenborg, L.; Brunner, H.G.; et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet. Med. 2017, 19, 1055–1063. [Google Scholar] [CrossRef] [Green Version]
- Shendure, J.; Porreca, G.J.; Reppas, N.B.; Lin, X.; McCutcheon, J.P.; Rosenbaum, A.M.; Wang, M.D.; Zhang, K.; Mitra, R.D.; Church, G.M. Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome. Science 2005, 309, 1728–1732. [Google Scholar] [CrossRef] [Green Version]
- Braslavsky, I.; Hebert, B.; Kartalov, E.; Quake, S.R. Sequence information can be obtained from single DNA molecules. Proc. Natl. Acad. Sci. USA 2003, 100, 3960–3964. [Google Scholar] [CrossRef] [Green Version]
- Hintzsche, J.D.; Gorden, N.T.; Amato, C.M.; Kim, J.; Wuensch, K.E.; Robinson, S.E.; Applegate, A.J.; Couts, K.L.; Medina, T.M.; Wells, K.R.; et al. Whole-exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res. 2017, 27, 189–199. [Google Scholar] [CrossRef]
- Vergara, I.A.; Mintoff, C.P.; Sandhu, S.; McIntosh, L.; Young, R.J.; Wong, S.Q.; Colebatch, A.; Cameron, D.L.; Kwon, J.L.; Wolfe, R.; et al. Evolution of late-stage metastatic melanoma is dominated by aneuploidy and whole genome doubling. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Abdel-Rahman, M.H.; Sample, K.M.; Pilarski, R.; Walsh, T.; Grosel, T.; Kinnamon, D.; Boru, G.; Massengill, J.B.; Schoenfield, L.; Kelly, B.; et al. Whole Exome Sequencing Identifies Candidate Genes Associated with Hereditary Predisposition to Uveal Melanoma. Ophthalmology 2019, 127, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Jing, C.; Chang, X.; Ding, D.; Han, T.; Yang, J.; Lu, Z.; Hu, X.; Liu, Z.; Wang, J.; et al. Mutational landscape of gastric cancer and clinical application of genomic profiling based on target next-generation sequencing. J. Transl. Med. 2019, 17, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiman, A.; Kikuchi, H.; Scocchia, D.; Smith, P.; Tsang, Y.W.; Snead, D.; Cree, I.A. Validation of an NGS mutation detection panel for melanoma. BMC Cancer 2017, 17, 150. [Google Scholar] [CrossRef] [Green Version]
- Newell, F.; Wilmott, J.S.; Johansson, P.A.; Nones, K.; Addala, V.; Mukhopadhyay, P.; Broit, N.; Amato, C.M.; Van Gulick, R.; Kazakoff, S.H.; et al. Whole-genome sequencing of acral melanoma reveals genomic complexity and diversity. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Usera, A.D.-D.; Lorenzo-Salazar, J.M.; Rubio-Rodríguez, L.A.; Muñoz-Barrera, A.; Guillen-Guio, B.; Marcelino-Rodríguez, I.; García-Olivares, V.; Mendoza-Alvarez, A.; Corrales, A.; Íñigo-Campos, A.; et al. Evaluation of Whole-Exome Enrichment Solutions: Lessons from the High-End of the Short-Read Sequencing Scale. J. Clin. Med. 2020, 9, 3656. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Govindaraj, G.M.; Edavazhippurath, A.; Faisal, N.; Bhoyar, R.C.; Gupta, V.; Uppuluri, R.; Manakkad, S.P.; Kashyap, A.; Kumar, A.; et al. Whole genome sequencing identifies novel structural variant in a large Indian family affected with X-linked agammaglobulinemia. PLoS ONE 2021, 16, e0254407. [Google Scholar] [CrossRef]
- Hou, Y.C.; Neidich, J.A.; Duncavage, E.J.; Spencer, D.H.; Schroeder, M.C. Clinical whole-genome sequencing in cancer diagnosis. Hum. Mutat. 2022, 43, 1519–1530. [Google Scholar] [CrossRef]
- Li, Y.; PCAWG Structural Variation Working Group; Roberts, N.D.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar] [CrossRef]
- Cosenza, M.R.; Rodriguez-Martin, B.; Korbel, J.O. Structural Variation in Cancer: Role, Prevalence, and Mechanisms. Annu. Rev. Genom. Hum. Genet. 2022, 23, 123–152. [Google Scholar] [CrossRef] [PubMed]
- Takai, E.; Nakamura, H.; Chiku, S.; Kubo, E.; Ohmoto, A.; Totoki, Y.; Shibata, T.; Higuchi, R.; Yamamoto, M.; Furuse, J.; et al. Whole-exome Sequencing Reveals New Potential Susceptibility Genes for Japanese Familial Pancreatic Cancer. Ann. Surg. 2020, 275, e652–e658. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Mao, R.; Ren, G.; Liu, X.; Zhang, Y.; Wang, J.; Wang, Y.; Li, M.; Qiu, Q.; Wang, L.; et al. Whole Exome Sequencing Identifies Putative Predictors of Recurrent Prostate Cancer with High Accuracy. OMICS A J. Integr. Biol. 2019, 23, 380–388. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Wu, J.; Liu, Y.; Kang, S. Whole-exome sequencing reveals potential germline and somatic mutations in 60 malignant ovarian germ cell tumors. Biol. Reprod. 2021, 105, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Skopelitou, D.; Miao, B.; Srivastava, A.; Kumar, A.; Kuświk, M.; Dymerska, D.; Paramasivam, N.; Schlesner, M.; Lubinski, J.; Hemminki, K.; et al. Whole Exome Sequencing Identifies APCDD1 and HDAC5 Genes as Potentially Cancer Predisposing in Familial Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 1837. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gu, X.; Li, Z.; Zheng, C.; Wang, Z.; Zhou, M.; Chen, Z.; Li, M.; Li, D.; Xiang, J. Whole-exome sequencing of rectal cancer identifies locally recurrent mutations in the Wnt pathway. Aging 2021, 13, 23262–23283. [Google Scholar] [CrossRef] [PubMed]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Wilmott, J.S.; Johansson, P.A.; Newell, F.; Waddell, N.; Ferguson, P.; Quek, C.; Patch, A.-M.; Nones, K.; Shang, P.; Pritchard, A.L.; et al. Whole genome sequencing of melanomas in adolescent and young adults reveals distinct mutation landscapes and the potential role of germline variants in disease susceptibility. Int. J. Cancer 2018, 144, 1049–1060. [Google Scholar] [CrossRef]
- Araújo, G.; Marinho, A.N.R.; Anaissi, A.K.; Vinasco-Sandoval, T.; Ribeiro-Dos-Santos, A.; Vidal, A.; De Araújo, G.S.; Demachki, S.; Ribeiro-Dos-Santos, Â. Whole mitochondrial genome sequencing highlights mitochondrial impact in gastric cancer. Sci. Rep. 2019, 9, 15716. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Niu, L.; Xiao, Z.; Zheng, C.; Shen, Y.; Shi, Y.; Han, X. Whole-genome sequencing of prostate cancer reveals novel mutation-driven processes and molecular subgroups. Life Sci. 2019, 254, 117218. [Google Scholar] [CrossRef]
- Mendelaar, P.A.J.; Smid, M.; van Riet, J.; Angus, L.; Labots, M.; Steeghs, N.; Hendriks, M.P.; Cirkel, G.A.; van Rooijen, J.M.; Tije, A.J.T.; et al. Whole genome sequencing of metastatic colorectal cancer reveals prior treatment effects and specific metastasis features. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Nair, S.V.; Madhulaxmi; Thomas, G.; Ankathil, R. Next-Generation Sequencing in Cancer. J. Maxillofac. Oral Surg. 2020, 20, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Kamps, R.; Brandão, R.D.; van den Bosch, B.J.; Paulussen, A.D.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification. Int. J. Mol. Sci. 2017, 18, 308. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, M.; Gabriel, S.; Getz, G. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 2010, 11, 685–696. [Google Scholar] [CrossRef]
- Nakagawa, H.; Wardell, C.P.; Furuta, M.; Taniguchi, H.; Fujimoto, A. Cancer whole-genome sequencing: Present and future. Oncogene 2015, 34, 5943–5950. [Google Scholar] [CrossRef]
- Hussen, B.M.; Abdullah, S.T.; Salihi, A.; Sabir, D.K.; Sidiq, K.R.; Rasul, M.F.; Hidayat, H.J.; Ghafouri-Fard, S.; Taheri, M.; Jamali, E. The emerging roles of NGS in clinical oncology and personalized medicine. Pathol. Res. Pract. 2022, 230, 153760. [Google Scholar] [CrossRef]
- Gagan, J.; Van Allen, E.M. Next-generation sequencing to guide cancer therapy. Genome Med. 2015, 7, 80. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- NIH. Cancer Genome Atlas. In Encyclopedia of Genetics, Genomics, Proteomics and Informatics; Rédei, G.P., Ed.; Springer: Dordrecht, The Netherlands, 2008; p. 265. ISBN 9781402067549. [Google Scholar]
- Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International Network of Cancer Genome Projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Trotman, J.; Armstrong, R.; Firth, H.; Trayers, C.; Watkins, J.; Allinson, K.; Jacques, T.S.; Nicholson, J.C.; Burke, G.A.A.; Ambrose, J.C.; et al. The NHS England 100,000 Genomes Project: Feasibility and utility of centralised genome sequencing for children with cancer. Br. J. Cancer 2022, 127, 137–144. [Google Scholar] [CrossRef]
- Turnbull, C. Introducing whole-genome sequencing into routine cancer care: The Genomics England 100,000 Genomes Project. Ann. Oncol. 2018, 29, 784–787. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Gupta, R.; Filipp, F.V. Cancer systems biology of TCGA SKCM: Efficient detection of genomic drivers in melanoma. Sci. Rep. 2015, 5, srep07857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Liu, M.; Huang, Y.; Wang, Z.; Wang, Y.; He, K.; Bai, R.; Ying, T.; Zheng, Y. Differential Gene Expression and Methylation Analysis of Melanoma in TCGA Database to Further Study the Expression Pattern of KYNU in Melanoma. J. Pers. Med. 2022, 12, 1209. [Google Scholar] [CrossRef] [PubMed]
- Scatena, C.; Murtas, D.; Tomei, S. Cutaneous Melanoma Classification: The Importance of High-Throughput Genomic Technologies. Front. Oncol. 2021, 11, 635488. [Google Scholar] [CrossRef] [PubMed]
- Ablain, J.; Al Mahi, A.; Rothschild, H.; Prasad, M.; Aires, S.; Yang, S.; Dokukin, M.E.; Xu, S.; Dang, M.; Sokolov, I.; et al. Loss of NECTIN1 triggers melanoma dissemination upon local IGF1 depletion. Nat. Genet. 2022, 1–14. [Google Scholar] [CrossRef]
- Scherrer, E.; Hair, G.; Mt-Isa, S.; Pereira, M.; Chan, G.; Shui, I.; Arumugam, P.; Zarowiecki, M.; Witkowska, K.; Rahim, T.; et al. 1136P Feasibility of linking the UK 100,000 genomes project and real-world evidence databases for a melanoma patient population. Ann. Oncol. 2020, 31, S760–S761. [Google Scholar] [CrossRef]
- Griffith, M.; Miller, C.; Griffith, O.; Krysiak, K.; Skidmore, Z.; Ramu, A.; Walker, J.R.; Dang, H.X.; Trani, L.; Larson, D.; et al. Optimizing Cancer Genome Sequencing and Analysis. Cell Syst. 2015, 1, 210–223. [Google Scholar] [CrossRef] [Green Version]
- Ku, C.S.; Cooper, D.N.; Roukos, D.H. Clinical relevance of cancer genome sequencing. World J. Gastroenterol. 2013, 19, 2011–2018. [Google Scholar] [CrossRef]
- Koboldt, D.C. Best practices for variant calling in clinical sequencing. Genome Med. 2020, 12, 1–13. [Google Scholar] [CrossRef]
- Chen, H.; Li, J.; Wang, Y.; Ng, P.K.-S.; Tsang, Y.H.; Shaw, K.R.; Mills, G.; Liang, H. Comprehensive assessment of computational algorithms in predicting cancer driver mutations. Genome Biol. 2020, 21, 1–17. [Google Scholar] [CrossRef]
- Borad, M.J.; Egan, J.B.; Condjella, R.M.; Liang, W.S.; Fonseca, R.; Ritacca, N.R.; McCullough, A.E.; Barrett, M.T.; Hunt, K.S.; Champion, M.D.; et al. Clinical Implementation of Integrated Genomic Profiling in Patients with Advanced Cancers. Sci. Rep. 2016, 6, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, K.; Shah, M.; Johnson, M.; Sanghvi, R.; Shelton, J.; Nagulapalli, K.; Oschwald, D.M.; Zody, M.C.; Germer, S.; Jobanputra, V.; et al. Deep whole-genome sequencing of 3 cancer cell lines on 2 sequencing platforms. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, M.; Juhos, S.; Larsson, M.; Olason, P.I.; Martin, M.; Eisfeldt, J.; DiLorenzo, S.; Sandgren, J.; De Ståhl, T.D.; Ewels, P.; et al. Sarek: A portable workflow for whole-genome sequencing analysis of germline and somatic variants. F1000Research 2020, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2020; ISBN 9781491975169. [Google Scholar]
- Nakagawa, H.; Fujita, M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018, 109, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köster, J.; Rahmann, S. Snakemake--a scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss, K.; Van der Auwera, G.; Gentry, J. Full-stack genomics pipelining with GATK4 + WDL + Cromwell. F1000Research 2017, 6. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Li, H.; Dawood, M.; Khayat, M.M.; Farek, J.R.; Jhangiani, S.N.; Khan, Z.M.; Mitani, T.; Coban-Akdemir, Z.; Lupski, J.R.; Venner, E.; et al. Exome variant discrepancies due to reference-genome differences. Am. J. Hum. Genet. 2021, 108, 1239–1250. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Aganezov, S.; Yan, S.M.; Soto, D.C.; Kirsche, M.; Zarate, S.; Avdeyev, P.; Taylor, D.J.; Shafin, K.; Shumate, A.; Xiao, C.; et al. A complete reference genome improves analysis of human genetic variation. Science 2022, 376, eabl3533. [Google Scholar] [CrossRef]
- Alkan, C.; Carbone, L.; Dennis, M.; Ernst, J.; Evrony, G.; Girirajan, S.; Leung, D.C.Y.; Cheng, C.C.; MacAlpine, D.; Ni, T.; et al. Implications of the first complete human genome assembly. Genome Res. 2022, 32, 595–598. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Mu, J.C.; Jiang, H.; Kiani, A.; Mohiyuddin, M.; Asadi, N.B.; Wong, W.H. Fast and accurate read alignment for resequencing. Bioinformatics 2012, 28, 2366–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [Green Version]
- Broad Institute. Picard Toolkit. Available online: http://broadinstitute.github.io/picard/ (accessed on 10 October 2022).
- Pedersen, B.S.; Quinlan, A.R. Mosdepth: Quick Coverage Calculation for Genomes and Exomes. Bioinformatics 2018, 34, 867–868. [Google Scholar] [CrossRef] [Green Version]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, D.; Sato, T.; Cibulskis, K.; Getz, G.; Stewart, C.; Lichtenstein, L. Calling Somatic SNVs and Indels with Mutect2. BioRxiv 2019, 861054. [Google Scholar] [CrossRef]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Patiyal, S.; Dhall, A.; Raghava, G.P.S. Prediction of risk-associated genes and high-risk liver cancer patients from their mutation profile: Benchmarking of mutation calling techniques. Biol. Methods Protoc. 2022, 7, bpac012. [Google Scholar] [CrossRef]
- Zhou, H.; Hu, Y.; Luo, R.; Zhao, Y.; Pan, H.; Ji, L.; Zhou, T.; Zhang, L.; Long, H.; Fu, J.; et al. Multi-region exome sequencing reveals the intratumoral heterogeneity of surgically resected small cell lung cancer. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Ura, H.; Togi, S.; Niida, Y. Dual Deep Sequencing Improves the Accuracy of Low-Frequency Somatic Mutation Detection in Cancer Gene Panel Testing. Int. J. Mol. Sci. 2020, 21, 3530. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Markovets, A.; Ahdesmäki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Z.; Brosnan, M.; Sokol, E.S.; Xie, M.; Dry, J.R.; Harrington, E.A.; Barrett, J.C.; Hodgson, D. Landscape of homologous recombination deficiencies in solid tumours: Analyses of two independent genomic datasets. BMC Cancer 2022, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2015, 12, e1004873. [Google Scholar] [CrossRef] [Green Version]
- Vittoria, M.A.; Kingston, N.; Kotynkova, K.; Xia, E.; Hong, R.; Huang, L.; McDonald, S.; Tilston-Lunel, A.; Darp, R.; Campbell, J.D.; et al. Inactivation of the Hippo tumor suppressor pathway promotes melanoma. Nat. Commun. 2022, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Zhang, Z.; Dong, X.; Yang, R.; Duan, Z.; Xiang, Z.; Li, J.; Li, G.; Yan, F.; Xue, H.; et al. Pangenomic analysis of Chinese gastric cancer. Nat. Commun. 2022, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.; Kim, H.; Min, K.I.; Kim, C.; Kwon, Y.; Zheng, Z.; Kim, Y.; Park, H.-S.; Ju, Y.S.; Roh, M.R.; et al. Weight-bearing activity impairs nuclear membrane and genome integrity via YAP activation in plantar melanoma. Nat. Commun. 2022, 13, 1–15. [Google Scholar] [CrossRef]
- Layer, R.M.; Chiang, C.; Quinlan, A.R.; Hall, I.M. LUMPY: A probabilistic framework for structural variant discovery. Genome Biol. 2014, 15, R84. [Google Scholar] [CrossRef] [Green Version]
- Akdemir, K.C.; Le, V.T.; Chandran, S.; Li, Y.; Verhaak, R.G.; Beroukhim, R.; Campbell, P.J.; Chin, L.; Dixon, J.R.; Futreal, P.A.; et al. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat. Genet. 2020, 52, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Cameron, D.L.; Baber, J.; Shale, C.; Valle-Inclan, J.E.; Besselink, N.; van Hoeck, A.; Janssen, R.; Cuppen, E.; Priestley, P.; Papenfuss, A.T. GRIDSS2: Comprehensive characterisation of somatic structural variation using single breakend variants and structural variant phasing. Genome Biol. 2021, 22, 1–25. [Google Scholar] [CrossRef]
- Tiong, M.I.S.; Wilson, B.C.; Yerneni, M.S.; Markham, J.; Dun, B.K.; Bajel, F.A.; Thompson, B.E.R.; Westerman, M.D.A.; Blombery, M.P. Mutational and Copy Number Profiling of Circulating Tumor DNA in Acute Myeloid Leukemia Using Targeted Next Generation Sequencing. Blood 2020, 136, 39–40. [Google Scholar] [CrossRef]
- Field, M.G.; Durante, M.A.; Anbunathan, H.; Cai, L.Z.; Decatur, C.L.; Bowcock, A.M.; Kurtenbach, S.; Harbour, J.W. Punctuated evolution of canonical genomic aberrations in uveal melanoma. Nat. Commun. 2018, 9, 116. [Google Scholar] [CrossRef] [Green Version]
- Demidov, G.; Ossowski, S. ClinCNV: Novel Method for Allele-Specific Somatic Copy-Number Alterations Detection. bioRxiv 2019, 837971. [Google Scholar] [CrossRef]
- Prasad, A.; Rabionet, R.; Espinet, B.; Zapata, L.; Puiggros, A.; Melero, C.; Puig, A.; Sarria-Trujillo, Y.; Ossowski, S.; Garcia-Muret, M.P.; et al. Identification of Gene Mutations and Fusion Genes in Patients with Sézary Syndrome. J. Investig. Dermatol. 2016, 136, 1490–1499. [Google Scholar] [CrossRef] [Green Version]
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.; Stebbings, E.; Grigoriadou, S.; Wood, N.; Hambleton, S.; Burns, S.; Thrasher, A.; et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Boujemaa, M.; Hamdi, Y.; Mejri, N.; Romdhane, L.; Ghedira, K.; Bouaziz, H.; El Benna, H.; Labidi, S.; Dallali, H.; Jaidane, O.; et al. Germline copy number variations in BRCA1/2 negative families: Role in the molecular etiology of hereditary breast cancer in Tunisia. PLoS ONE 2021, 16, e0245362. [Google Scholar] [CrossRef]
- Minoche, A.E.; Ben Lundie, B.; Peters, G.B.; Ohnesorg, T.; Pinese, M.; Thomas, D.M.; Zankl, A.; Roscioli, T.; Schonrock, N.; Kummerfeld, S.; et al. ClinSV: Clinical grade structural and copy number variant detection from whole genome sequencing data. Genome Med. 2021, 13, 1–19. [Google Scholar] [CrossRef]
- Deng, N.; Minoche, A.; Harvey, K.; Li, M.; Winkler, J.; Goga, A.; Swarbrick, A. Deep whole genome sequencing identifies recurrent genomic alterations in commonly used breast cancer cell lines and patient-derived xenograft models. Breast Cancer Res. 2022, 24, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Prieto, C.A.; Martínez-Jiménez, F.; Valencia, A.; Porta-Pardo, E. Detection of oncogenic and clinically actionable mutations in cancer genomes critically depends on variant calling tools. Bioinformatics 2022, 38, 3181–3191. [Google Scholar] [CrossRef]
- Xu, C. A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Comput. Struct. Biotechnol. J. 2018, 16, 15–24. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Lv, N.; Liao, J.; Long, J.; Xue, R.; Ai, N.; Xu, D.; Fan, X. Copy number variation is highly correlated with differential gene expression: A pan-cancer study. BMC Med. Genet. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Cho, S. Set-Wise Differential Interaction Between Copy Number Alterations and Gene Expressions of Lower-Grade Glioma Reveals Prognosis-Associated Pathways. Entropy 2020, 22, 1434. [Google Scholar] [CrossRef]
- Shahrisa, A.; Tahmasebi-Birgani, M.; Ansari, H.; Mohammadi, Z.; Carloni, V.; Asl, J.M. The pattern of gene copy number alteration (CNAs) in hepatocellular carcinoma: An in silico analysis. Mol. Cytogenet. 2021, 14, 1–10. [Google Scholar] [CrossRef]
- van Belzen, I.A.E.M.; Schönhuth, A.; Kemmeren, P.; Hehir-Kwa, J.Y. Structural variant detection in cancer genomes: Computational challenges and perspectives for precision oncology. Npj Precis. Oncol. 2021, 5, 1–11. [Google Scholar] [CrossRef]
- Gong, T.; Hayes, V.M.; Chan, E.K.F. Detection of somatic structural variants from short-read next-generation sequencing data. Brief. Bioinform. 2020, 22, bbaa056. [Google Scholar] [CrossRef]
- Cameron, D.L.; Di Stefano, L.; Papenfuss, A.T. Comprehensive evaluation and characterisation of short read general-purpose structural variant calling software. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Coutelier, M.; Holtgrewe, M.; Jäger, M.; Flöttman, R.; Mensah, M.A.; Spielmann, M.; Krawitz, P.; Horn, D.; Beule, D.; Mundlos, S. Combining callers improves the detection of copy number variants from whole-genome sequencing. Eur. J. Hum. Genet. 2021, 30, 178–186. [Google Scholar] [CrossRef]
- Olson, N.D.; Wagner, J.; McDaniel, J.; Stephens, S.H.; Westreich, S.T.; Prasanna, A.G.; Johanson, E.; Boja, E.; Maier, E.J.; Serang, O.; et al. PrecisionFDA Truth Challenge V2: Calling variants from short and long reads in difficult-to-map regions. Cell Genom. 2022, 2, 100129. [Google Scholar] [CrossRef]
- Wagner, J.; Olson, N.D.; Harris, L.; McDaniel, J.; Cheng, H.; Fungtammasan, A.; Hwang, Y.-C.; Gupta, R.; Wenger, A.M.; Rowell, W.J.; et al. Curated variation benchmarks for challenging medically relevant autosomal genes. Nat. Biotechnol. 2022, 40, 672–680. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [Green Version]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Dashti, M.J.S.; Gamieldien, J. A practical guide to filtering and prioritizing genetic variants. BioTechniques 2017, 62, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Belyeu, J.R.; Chowdhury, M.; Brown, J.; Pedersen, B.S.; Cormier, M.J.; Quinlan, A.R.; Layer, R.M. Samplot: A platform for structural variant visual validation and automated filtering. Genome Biol. 2021, 22, 1–13. [Google Scholar] [CrossRef]
- Liu, Z.; Roberts, R.; Mercer, T.R.; Xu, J.; Sedlazeck, F.J.; Tong, W. Towards accurate and reliable resolution of structural variants for clinical diagnosis. Genome Biol. 2022, 23, 1–25. [Google Scholar] [CrossRef]
- Griffith, M.; Spies, N.C.; Krysiak, K.; McMichael, J.F.; Coffman, A.C.; Danos, A.M.; Ainscough, B.J.; Ramirez, C.A.; Rieke, D.T.; Kujan, L.; et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet. 2017, 49, 170–174. [Google Scholar] [CrossRef] [Green Version]
- Ainscough, B.J.; Barnell, E.K.; Ronning, P.; Campbell, K.M.; Wagner, A.H.; Fehniger, T.A.; Dunn, G.P.; Uppaluri, R.; Govindan, R.; Rohan, T.E.; et al. A deep learning approach to automate refinement of somatic variant calling from cancer sequencing data. Nat. Genet. 2018, 50, 1735–1743. [Google Scholar] [CrossRef]
- Vaisband, M.; Schubert, M.; Gassner, F.J.; Geisberger, R.; Greil, R.; Zaborsky, N.; Hasenauer, J. Validation of Genetic Variants from NGS Data Using Deep Convolutional Neural Networks. bioRxiv 2022, 488021. [Google Scholar] [CrossRef]
- Myers, M.A.; Zaccaria, S.; Raphael, B.J. Identifying tumor clones in sparse single-cell mutation data. Bioinformatics 2020, 36, i186–i193. [Google Scholar] [CrossRef]
- Zhang, X.; Lv, D.; Zhang, Y.; Liu, Q.; Li, Z. Clonal evolution of acute myeloid leukemia highlighted by latest genome sequencing studies. Oncotarget 2016, 7, 58586–58594. [Google Scholar] [CrossRef] [Green Version]
- Strom, S.P. Current practices and guidelines for clinical next-generation sequencing oncology testing. Cancer Biol. Med. 2016, 13, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [Green Version]
- Gatalica, Z.; Xiu, J.; Swensen, J.; Vranic, S. Molecular characterization of cancers with NTRK gene fusions. Mod. Pathol. 2018, 32, 147–153. [Google Scholar] [CrossRef]
- Quan, V.L.; Panah, E.; Zhang, B.; Shi, K.; Mohan, L.S.; Gerami, P. The role of gene fusions in melanocytic neoplasms. J. Cutan. Pathol. 2019, 46, 878–887. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-F.; Wang, W.-X.; Xu, C.-W.; Huang, L.-C.; Li, X.-F.; Lan, G.; Zhai, Z.-Q.; Zhu, Y.-C.; Du, K.-Q.; Lei, L.; et al. A novel SOS1-ALK fusion variant in a patient with metastatic lung adenocarcinoma and a remarkable response to crizotinib. Lung Cancer 2020, 142, 59–62. [Google Scholar] [CrossRef]
- Mittal, V.K.; McDonald, J.F. De novo assembly and characterization of breast cancer transcriptomes identifies large numbers of novel fusion-gene transcripts of potential functional significance. BMC Med. Genom. 2017, 10, 1–20. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Krueger, F.; James, F.; Ewels, P.; Afyounian, E.; Schuster-Boeckler, B. TrimGalore: V0.6.7—DOI via Zenodo. 2021. Available online: https://zenodo.org/record/5127899 (accessed on 15 October 2022).
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Robertson, G.; Schein, J.; Chiu, R.; Corbett, R.; Field, M.; Jackman, S.D.; Mungall, K.; Lee, S.; Okada, H.M.; Qian, J.Q.; et al. De novo assembly and analysis of RNA-seq data. Nat. Methods 2010, 7, 909–912. [Google Scholar] [CrossRef]
- Chiu, R.; Nip, K.M.; Chu, J.; Birol, I. TAP: A targeted clinical genomics pipeline for detecting transcript variants using RNA-seq data. BMC Med. Genom. 2018, 11, 79. [Google Scholar] [CrossRef]
- Raghavan, V.; Kraft, L.; Mesny, F.; Rigerte, L. A simple guide to de novo transcriptome assembly and annotation. Brief. Bioinform. 2022, 23, bbab563. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Chiu, R.; Nip, K.M.; Birol, I. Fusion-Bloom: Fusion detection in assembled transcriptomes. Bioinformatics 2019, 36, 2256–2257. [Google Scholar] [CrossRef]
- Zhang, J.; White, N.M.; Schmidt, H.K.; Fulton, R.S.; Tomlinson, C.; Warren, W.C.; Wilson, R.K.; Maher, C.A. INTEGRATE: Gene fusion discovery using whole genome and transcriptome data. Genome Res. 2015, 26, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Endrullat, C.; Glökler, J.; Franke, P.; Frohme, M. Standardization and quality management in next-generation sequencing. Appl. Transl. Genom. 2016, 10, 2–9. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data 2010. Available online: https://www.scienceopen.com/document?vid=de674375-ab83-4595-afa9-4c8aa9e4e736 (accessed on 15 October 2022).
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.-M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef] [Green Version]
- Thornton, T.; Tang, H.; Hoffmann, T.J.; Ochs-Balcom, H.M.; Caan, B.J.; Risch, N. Estimating Kinship in Admixed Populations. Am. J. Hum. Genet. 2012, 91, 122–138. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Chen, L. Inference of kinship using spatial distributions of SNPs for genome-wide association studies. BMC Genom. 2016, 17, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.S.; Bhetariya, P.J.; Brown, J.; Kravitz, S.N.; Marth, G.; Jensen, R.L.; Bronner, M.P.; Underhill, H.R.; Quinlan, A.R. Somalier: Rapid relatedness estimation for cancer and germline studies using efficient genome sketches. Genome Med. 2020, 12, 1–9. [Google Scholar] [CrossRef]
- Webster, T.H.; Couse, M.; Grande, B.M.; Karlins, E.; Phung, T.N.; Richmond, P.A.; Whitford, W.; Wilson Sayres, M.A. Identifying, Understanding, and Correcting Technical Biases on the Sex Chromosomes in next-Generation Sequencing Data. bioRxiv 2018, 346940. [Google Scholar] [CrossRef] [Green Version]
- Genomics Division, ITER SexQC-for-NGS-Data: Sex Quality Control for Next Generation Sequencing Data; Github. Available online: https://github.com/genomicsITER/sexQC-for-NGS-data (accessed on 15 October 2022).
- Pollard, M.O.; Gurdasani, D.; Mentzer, A.J.; Porter, T.; Sandhu, M.S. Long reads: Their purpose and place. Hum. Mol. Genet. 2018, 27, R234–R241. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, Y.; Sereewattanawoot, S.; Suzuki, A. A new era of long-read sequencing for cancer genomics. J. Hum. Genet. 2019, 65, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.C.; Bell, J.M.; Wood-Bouwens, C.; Chen, J.J.; Zhang, N.R.; Ji, H.P. Identification of large rearrangements in cancer genomes with barcode linked reads. Nucleic Acids Res. 2017, 46, e19. [Google Scholar] [CrossRef] [Green Version]
- Dozmorov, M.G.; Tyc, K.M.; Sheffield, N.C.; Boyd, D.C.; Olex, A.L.; Reed, J.; Harrell, J.C. Chromatin conformation capture (Hi-C) sequencing of patient-derived xenografts: Analysis guidelines. GigaScience 2021, 10, giab022. [Google Scholar] [CrossRef]
- Chan, E.K.; Cameron, D.L.; Petersen, D.C.; Lyons, R.J.; Baldi, B.F.; Papenfuss, A.T.; Thomas, D.M.; Hayes, V.M. Optical mapping reveals a higher level of genomic architecture of chained fusions in cancer. Genome Res. 2018, 28, 726–738. [Google Scholar] [CrossRef]
- Euskirchen, P.; Bielle, F.; Labreche, K.; Kloosterman, W.P.; Rosenberg, S.; Daniau, M.; Schmitt, C.; Masliah-Planchon, J.; Bourdeaut, F.; Dehais, C.; et al. Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathol. 2017, 134, 691–703. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, Y.; Miyake, S.; Oka, M.; Kanai, A.; Kawai, Y.; Nagasawa, S.; Shiraishi, Y.; Tokunaga, K.; Kohno, T.; Seki, M.; et al. Phasing analysis of lung cancer genomes using a long read sequencer. Nat. Commun. 2022, 13, 1–17. [Google Scholar] [CrossRef]
- Viswanathan, S.R.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.W.; Haradhvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoades, J.; et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 2018, 174, 433–447.e19. [Google Scholar] [CrossRef] [Green Version]
- Greer, S.U.; Nadauld, L.D.; Lau, B.T.; Chen, J.; Wood-Bouwens, C.; Ford, J.M.; Kuo, C.J.; Ji, H.P. Linked read sequencing resolves complex genomic rearrangements in gastric cancer metastases. Genome Med. 2017, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ren, B.; Yang, J.; Wang, C.; Yang, G.; Wang, H.; Chen, Y.; Xu, R.; Fan, X.; You, L.; Zhang, T.; et al. High-resolution Hi-C maps highlight multiscale 3D epigenome reprogramming during pancreatic cancer metastasis. J. Hematol. Oncol. 2021, 14, 1–19. [Google Scholar] [CrossRef]
- Suttorp, J.; Lühmann, J.L.; Behrens, Y.L.; Göhring, G.; Steinemann, D.; Reinhardt, D.; von Neuhoff, N.; Schneider, M. Optical Genome Mapping as a Diagnostic Tool in Pediatric Acute Myeloid Leukemia. Cancers 2022, 14, 2058. [Google Scholar] [CrossRef]
- Magi, A.; Semeraro, R.; Mingrino, A.; Giusti, B.; D’Aurizio, R. Nanopore sequencing data analysis: State of the art, applications and challenges. Brief. Bioinform. 2017, 19, 1256–1272. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Rang, F.J.; Kloosterman, W.P.; De Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Ott, A.; Schnable, J.C.; Yeh, C.T.; Wu, L.; Liu, C.; Hu, H.C.; Dalgard, C.L.; Sarkar, S.; Schnable, P.S. Linked read technology for assembling large complex and polyploid genomes. BMC Genom. 2018, 19, 651. [Google Scholar] [CrossRef]
- Van Berkum, N.L.; Lieberman-Aiden, E.; Williams, L.; Imakaev, M.; Gnirke, A.; Mirny, L.A.; Dekker, J.; Lander, E.S. Hi-C: A Method to Study the Three-dimensional Architecture of Genomes. J. Vis. Exp. 2010, 39, e1869. [Google Scholar] [CrossRef] [Green Version]
- Yardımcı, G.G.; Ozadam, H.; Sauria, M.E.G.; Ursu, O.; Yan, K.-K.; Yang, T.; Chakraborty, A.; Kaul, A.; Lajoie, B.R.; Song, F.; et al. Measuring the reproducibility and quality of Hi-C data. Genome Biol. 2019, 20, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Kyriakidou, M.; Tai, H.H.; Anglin, N.L.; Ellis, D.; Strömvik, M.V. Current Strategies of Polyploid Plant Genome Sequence Assembly. Front. Plant Sci. 2018, 9, 1660. [Google Scholar] [CrossRef]
- Oluwadare, O.; Highsmith, M.; Cheng, J. An Overview of Methods for Reconstructing 3-D Chromosome and Genome Structures from Hi-C Data. Biol. Proced. Online 2019, 21, 7. [Google Scholar] [CrossRef]
- Shi, L.; Guo, Y.; Dong, C.; Huddleston, J.; Yang, H.; Han, X.; Fu, A.; Li, Q.; Li, N.; Gong, S.; et al. Long-read sequencing and de novo assembly of a Chinese genome. Nat. Commun. 2016, 7, 12065. [Google Scholar] [CrossRef] [Green Version]
- Sahajpal, N.S.; Lai, C.-Y.J.; Hastie, A.; Mondal, A.K.; Dehkordi, S.R.; van der Made, C.I.; Fedrigo, O.; Al-Ajli, F.; Jalnapurkar, S.; Byrska-Bishop, M.; et al. Optical genome mapping identifies rare structural variations as predisposition factors associated with severe COVID-19. iScience 2022, 25, 103760. [Google Scholar] [CrossRef]
- Goldrich, D.; LaBarge, B.; Chartrand, S.; Zhang, L.; Sadowski, H.; Zhang, Y.; Pham, K.; Way, H.; Lai, C.-Y.; Pang, A.; et al. Identification of Somatic Structural Variants in Solid Tumors by Optical Genome Mapping. J. Pers. Med. 2021, 11, 142. [Google Scholar] [CrossRef]
- Miga, K.H.; Koren, S.; Rhie, A.; Vollger, M.R.; Gershman, A.; Bzikadze, A.; Brooks, S.; Howe, E.; Porubsky, D.; Logsdon, G.A.; et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature 2020, 585, 79–84. [Google Scholar] [CrossRef]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, K.-T.; Slevin, M.K.; Meyerson, M.; Li, H. Identifying and correcting repeat-calling errors in nanopore sequencing of telomeres. Genome Biol. 2022, 23, 1–16. [Google Scholar] [CrossRef]
- Jones, A.; Torkel, C.; Stanley, D.; Nasim, J.; Borevitz, J.; Schwessinger, B. High-molecular weight DNA extraction, clean-up and size selection for long-read sequencing. PLoS ONE 2021, 16, e0253830. [Google Scholar] [CrossRef]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Shumate, A.; Zimin, A.V.; Sherman, R.M.; Puiu, D.; Wagner, J.M.; Olson, N.D.; Pertea, M.; Salit, M.L.; Zook, J.M.; Salzberg, S.L. Assembly and annotation of an Ashkenazi human reference genome. Genome Biol. 2020, 21, 1–18. [Google Scholar] [CrossRef]
- Xiao, C.; Chen, Z.; Chen, W.; Padilla, C.; Fang, L.-T.; Liu, T.; Schneider, V.; Wang, C.; Xiao, W. Personalized Genome Assembly for Accurate Cancer Somatic Mutation Discovery Using Cancer-Normal Paired Reference Samples. bioRxiv 2021, 438252. [Google Scholar] [CrossRef]
- Rosenfeld, J.A.; Mason, C.E.; Smith, T.M. Limitations of the Human Reference Genome for Personalized Genomics. PLoS ONE 2012, 7, e40294. [Google Scholar] [CrossRef] [PubMed]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leger, A.; Leonardi, T. pycoQC, interactive quality control for Oxford Nanopore Sequencing. J. Open Source Softw. 2019, 4. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Au, K.F. The blooming of long-read sequencing reforms biomedical research. Genome Biol. 2022, 23, 1–4. [Google Scholar] [CrossRef]
- Ccs: CCS: Generate Highly Accurate Single-Molecule Consensus Reads (HiFi Reads); Github. Available online: https://github.com/PacificBiosciences/ccs (accessed on 15 October 2022).
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Salmela, L.; Walve, R.M.; Rivals, E.; Ukkonen, E. Accurate self-correction of errors in long reads using de Bruijn graphs. Bioinformatics 2016, 33, 799–806. [Google Scholar] [CrossRef] [Green Version]
- Snajder, R.; Leger, A.; Stegle, O.; Bonder, M.J. PycoMeth: A Toolbox for Differential Methylation Testing from Nanopore Methylation Calls. bioRxiv 2022, 480699. [Google Scholar] [CrossRef]
- Ni, P.; Huang, N.; Zhang, Z.; Wang, D.-P.; Liang, F.; Miao, Y.; Xiao, C.-L.; Luo, F.; Wang, J. DeepSignal: Detecting DNA methylation state from Nanopore sequencing reads using deep-learning. Bioinformatics 2019, 35, 4586–4595. [Google Scholar] [CrossRef]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Edge, P.; Bansal, V. Longshot enables accurate variant calling in diploid genomes from single-molecule long read sequencing. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Shafin, K.; Pesout, T.; Chang, P.-C.; Nattestad, M.; Kolesnikov, A.; Goel, S.; Baid, G.; Kolmogorov, M.; Eizenga, J.M.; Miga, K.H.; et al. Haplotype-aware variant calling with PEPPER-Margin-DeepVariant enables high accuracy in nanopore long-reads. Nat. Methods 2021, 18, 1322–1332. [Google Scholar] [CrossRef]
- Heller, D.; Vingron, M. SVIM: Structural variant identification using mapped long reads. Bioinformatics 2019, 35, 2907–2915. [Google Scholar] [CrossRef] [Green Version]
- Heller, D.; Vingron, M. SVIM-asm: Structural variant detection from haploid and diploid genome assemblies. Bioinformatics 2020, 36, 5519–5521. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Liu, Y.; Jiang, Y.; Li, J.; Gao, Y.; Cui, Z.; Liu, Y.; Liu, B.; Wang, Y. Long-read-based human genomic structural variation detection with cuteSV. Genome Biol. 2020, 21, 1–24. [Google Scholar] [CrossRef]
- Freire, B.; Ladra, S.; Parama, J.R. Memory-Efficient Assembly using Flye. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021; online ahead of print. [Google Scholar] [CrossRef]
- Shafin, K.; Pesout, T.; Lorig-Roach, R.; Haukness, M.; Olsen, H.E.; Bosworth, C.; Armstrong, J.; Tigyi, K.; Maurer, N.; Koren, S.; et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol. 2020, 38, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA genome assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [Green Version]
- Di Genova, A.; Buena-Atienza, E.; Ossowski, S.; Sagot, M.-F. Efficient hybrid de novo assembly of human genomes with WENGAN. Nat. Biotechnol. 2020, 39, 422–430. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Belser, C.; Istace, B.; Denis, E.; Dubarry, M.; Baurens, F.-C.; Falentin, C.; Genete, M.; Berrabah, W.; Chèvre, A.-M.; Delourme, R.; et al. Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps. Nat. Plants 2018, 4, 879–887. [Google Scholar] [CrossRef]
- Ballouz, S.; Dobin, A.; Gillis, J.A. Is it time to change the reference genome? Genome Biol. 2019, 20, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valiente-Mullor, C.; Beamud, B.; Ansari, I.; Francés-Cuesta, C.; García-González, N.; Mejía, L.; Ruiz-Hueso, P.; González-Candelas, F. One is not enough: On the effects of reference genome for the mapping and subsequent analyses of short-reads. PLoS Comput. Biol. 2021, 17, e1008678. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Jeon, S.; Kim, C.; Kim, Y.K.; Cho, Y.S.; Kim, J.; Blazyte, A.; Manica, A.; Lee, S.; Bhak, J. Chromosome-scale assembly comparison of the Korean Reference Genome KOREF from PromethION and PacBio with Hi-C mapping information. GigaScience 2019, 8, giz125. [Google Scholar] [CrossRef] [PubMed]
- Ouzhuluobu; He, Y.; Lou, H.; Cui, C.; Deng, L.; Gao, Y.; Zheng, W.; Guo, Y.; Wang, X.; Ning, Z.; et al. De novo assembly of a Tibetan genome and identification of novel structural variants associated with high-altitude adaptation. Natl. Sci. Rev. 2019, 7, 391–402. [Google Scholar] [CrossRef]
- Li, Z.; Chen, Y.; Mu, D.; Yuan, J.; Shi, Y.; Zhang, H.; Gan, J.; Li, N.; Hu, X.; Liu, B.; et al. Comparison of the two major classes of assembly algorithms: Overlap-layout-consensus and de-bruijn-graph. Brief. Funct. Genom. 2011, 11, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.R.; Pervez, M.T.; Babar, M.E.; Naveed, N.; Shoaib, M. A Comprehensive Study of De Novo Genome Assemblers: Current Challenges and Future Prospective. Evol. Bioinform. 2018, 14, 1176934318758650. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Erickson, D.L.; Meng, J. Benchmarking hybrid assembly approaches for genomic analyses of bacterial pathogens using Illumina and Oxford Nanopore sequencing. BMC Genom. 2020, 21, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Li, T.; Vermeesch, J.R.; Xie, Z. A benchmark of structural variation detection by long reads through a realistic simulated model. Genome Biol. 2021, 22, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Abel, H.J.; Genomics, N.C.F.C.D.; Larson, D.E.; Regier, A.A.; Chiang, C.; Das, I.; Kanchi, K.L.; Layer, R.M.; Neale, B.M.; Salerno, W.J.; et al. Mapping and characterization of structural variation in 17,795 human genomes. Nature 2020, 583, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Jia, P.; Wang, S.; Ye, K. Comparison and Benchmark of Long-Read Based Structural Variant Detection Strategies. bioRxiv 2022, 503274. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Xu, L.; Seki, M.; Yokoyama, T.T.; Kasahara, M.; Kashima, Y.; Ohashi, A.; Shimada, Y.; Motoi, N.; Tsuchihara, K.; et al. Long-read sequencing for non-small-cell lung cancer genomes. Genome Res. 2020, 30, 1243–1257. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Koya, J.; Chiba, K.; Saito, Y.; Okada, A.; Kataoka, K. Precise Characterization of Somatic Structural Variations and Mobile Element Insertions from Paired Long-Read Sequencing Data with Nanomonsv. bioRxiv 2021, 214262. [Google Scholar] [CrossRef]
- Berger, M.F.; Mardis, E.R. The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 2018, 15, 353–365. [Google Scholar] [CrossRef]
- Mardis, E.R. The Impact of Next-Generation Sequencing on Cancer Genomics: From Discovery to Clinic. Cold Spring Harb. Perspect. Med. 2018, 9, a036269. [Google Scholar] [CrossRef]
- Cai, Z.; Poulos, R.C.; Liu, J.; Zhong, Q. Machine learning for multi-omics data integration in cancer. iScience 2022, 25, 7. [Google Scholar] [CrossRef]
- Wang, L.-B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528.e20. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Wong, J.H.; Yoshii, Y.; Akiyama, S.; Tanaka, A.; Yagi, H.; Shigemizu, D.; Nakagawa, H.; Mizokami, M.; Shimada, M. Whole-genome sequencing with long reads reveals complex structure and origin of structural variation in human genetic variations and somatic mutations in cancer. Genome Med. 2021, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]






| Brand | Instrument | Key Applications | Run Time (h) | Max. Output (Gb) | Max. Read Length (Bases) |
|---|---|---|---|---|---|
| Illumina, Inc. | NextSeq 550 | Targeted Gene Sequencing Transcriptome Sequencing | 12–30 | 120 | PE150 |
| NextSeq 1000 and 2000 | WGS (limited samples) WES Targeted Gene Sequencing Transcriptome Sequencing | 11–48 | 360 | PE150 | |
| NovaSeq 6000 | WGS WES Targeted Gene Sequencing Transcriptome Sequencing Methylation Sequencing | 13–44 * | 6000 | PE250 | |
| NovaSeq X Series | WGS (large sample number) WES Targeted Gene Sequencing Transcriptome Sequencing Methylation Sequencing | 13–48 * | 16,000 | PE150 | |
| MGI Tech | DNBSEQ-G50 | Targeted Gene Sequencing | 9–40 | 150 | PE150 |
| DNBSEQ-G400 | WGS (limited samples) WES Transcriptome Sequencing | 13–109 * | 1440 | PE300 | |
| DNBSEQ-T7 | WGS (large sample number) WES Targeted Gene Sequencing Transcriptome Sequencing | 24–30 | 6000 | PE150 | |
| Ion Torrent | Ion GeneStudio S5/Plus/Prime | WES (limited samples) Targeted Gene Sequencing | 6–19 | 15/30/50 | SE200/SE400/SE200 |
| Genexus System | WES (limited samples) Targeted Gene Sequencing | 2–3 | 15 | SE200 |
| Technology | Instruments | Read Characteristics | Related Somatic Studies |
|---|---|---|---|
| Oxford Nanopore Technologies | MinION GridION PromethION | Single molecule reads, average read length ~15–20 Kb (max ~2 Mb), with an error rate of 5–10% | Brain tumor [208], lung cancer [209] |
| Pacific Biosciences | Sequel Sequel II | HiFi reads, average read length ~15–20 Kb (max ~65 Kb), with error rate of 1% | Breast cancer [19] |
| Linked-reads (10x Genomics) | NextSeq HiSeq NovaSeq | Linked-reads obtained from short reads, average length ~100 Kb | Prostate cancer [210], gastric cancer [211] |
| Hi-C | NextSeq HiSeq NovaSeq | ~1 kb–1 Mb resolution, without base pair resolution | Pancreatic cancer [212] |
| Optical maps (BioNano Genomics) | NextSeq HiSeq NovaSeq | Optical mapping of long fragments, average length 250 Kb, without base pair resolution | Leukemia [213] |
| Bioinformatic Analysis | Tool | Sequencing Strategy | References |
|---|---|---|---|
| Base calling | Guppy, Bonito | ONT | https://github.com/nanoporetech/ (accessed on 2 August 2022) |
| Generate CCS | PacBio | [242] | |
| Quality control | pycoQC, NanoPack | ONT | [239,240] |
| Isoseq3 | PacBio | https://github.com/PacificBiosciences/IsoSeq (accessed on 2 August 2022) | |
| Read-error correction | Canu | ONT | [243] |
| LoRMA | PacBio | [244] | |
| DNA methylation | pycoMeth, DeepSignal, Megalodon | ONT | [245,246]; https://github.com/nanoporetech/megalodon (accessed on 2 August 2022) |
| pb-CpG-tools | PacBio | https://github.com/PacificBiosciences/pb-CpG-tools (accessed on 2 August 2022) | |
| Alignment | minimap2, NGMLR | ONT | [117,247] |
| pbmm2 | PacBio | https://github.com/PacificBiosciences/pbmm2 (accessed on 2 August 2022) | |
| SNV calling | Longshot, DeepVariant | ONT, PacBio | [248,249] |
| SV calling | Sniffles, SVIM, SVIM-asm, cuteSV | ONT | [247,250,251,252] |
| pbsv | PacBio | https://github.com/PacificBiosciences/pbsv (accessed on 2 August 2022) | |
| De novo assembly | Flye, Shasta | ONT | [253,254] |
| Hifiasm, FALCON | PacBio | [255,256] | |
| Hybrid assembly | MaSuRCA, WENGAN | ONT, PacBio | [257,258] |
| Polishing | Racon, Medaka, Pilon | ONT | [259,260] |
| Pilon, Quiver, Arrow | PacBio | [260,261] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz-Barrera, A.; Rubio-Rodríguez, L.A.; Díaz-de Usera, A.; Jáspez, D.; Lorenzo-Salazar, J.M.; González-Montelongo, R.; García-Olivares, V.; Flores, C. From Samples to Germline and Somatic Sequence Variation: A Focus on Next-Generation Sequencing in Melanoma Research. Life 2022, 12, 1939. https://doi.org/10.3390/life12111939
Muñoz-Barrera A, Rubio-Rodríguez LA, Díaz-de Usera A, Jáspez D, Lorenzo-Salazar JM, González-Montelongo R, García-Olivares V, Flores C. From Samples to Germline and Somatic Sequence Variation: A Focus on Next-Generation Sequencing in Melanoma Research. Life. 2022; 12(11):1939. https://doi.org/10.3390/life12111939
Chicago/Turabian StyleMuñoz-Barrera, Adrián, Luis A. Rubio-Rodríguez, Ana Díaz-de Usera, David Jáspez, José M. Lorenzo-Salazar, Rafaela González-Montelongo, Víctor García-Olivares, and Carlos Flores. 2022. "From Samples to Germline and Somatic Sequence Variation: A Focus on Next-Generation Sequencing in Melanoma Research" Life 12, no. 11: 1939. https://doi.org/10.3390/life12111939
APA StyleMuñoz-Barrera, A., Rubio-Rodríguez, L. A., Díaz-de Usera, A., Jáspez, D., Lorenzo-Salazar, J. M., González-Montelongo, R., García-Olivares, V., & Flores, C. (2022). From Samples to Germline and Somatic Sequence Variation: A Focus on Next-Generation Sequencing in Melanoma Research. Life, 12(11), 1939. https://doi.org/10.3390/life12111939