
Analysis of Signal Transduction Pathways Downstream M2 Receptor Activation: Effects on Schwann Cell Migration and Morphology

 and
and 
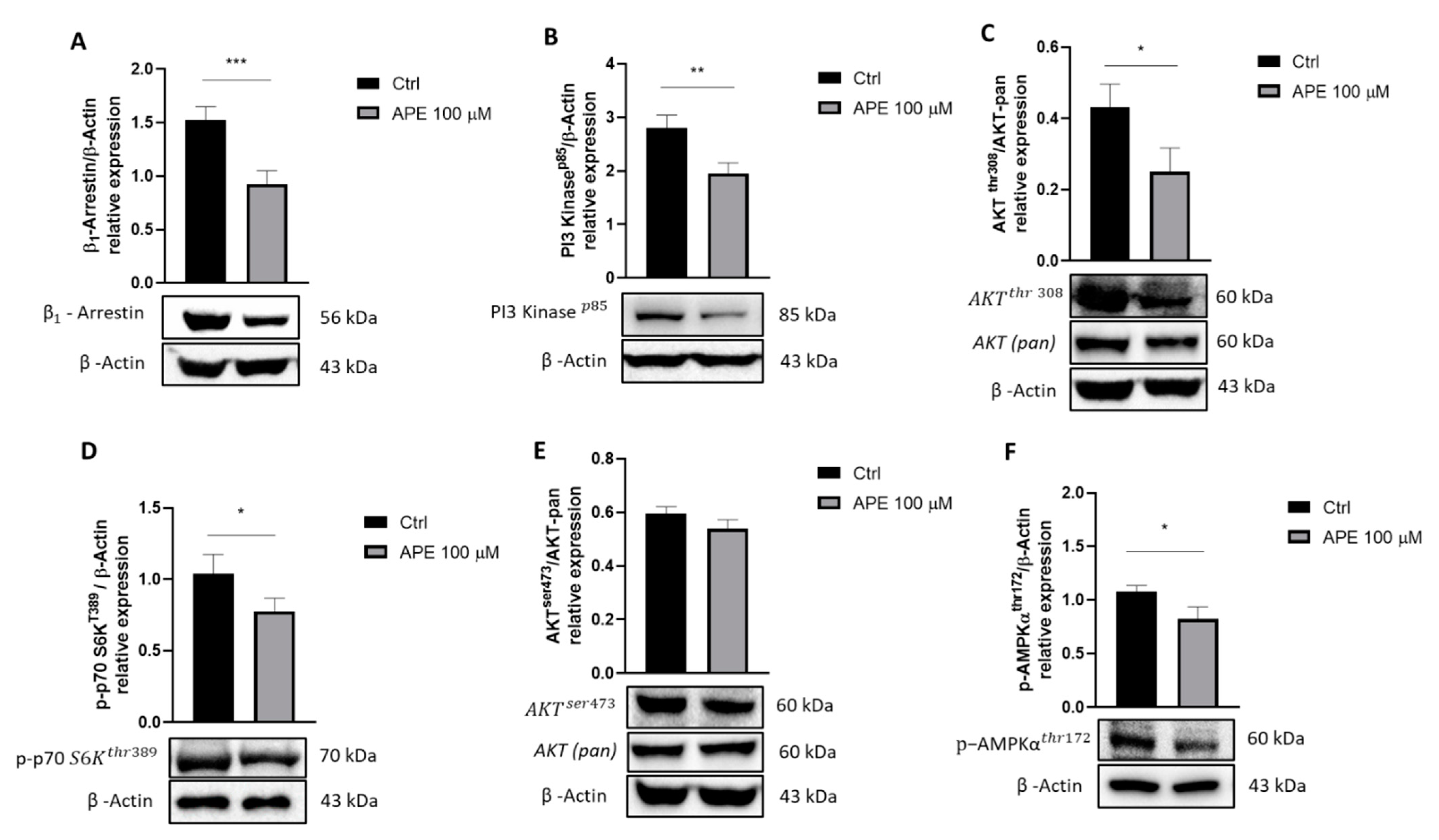{kind=link}
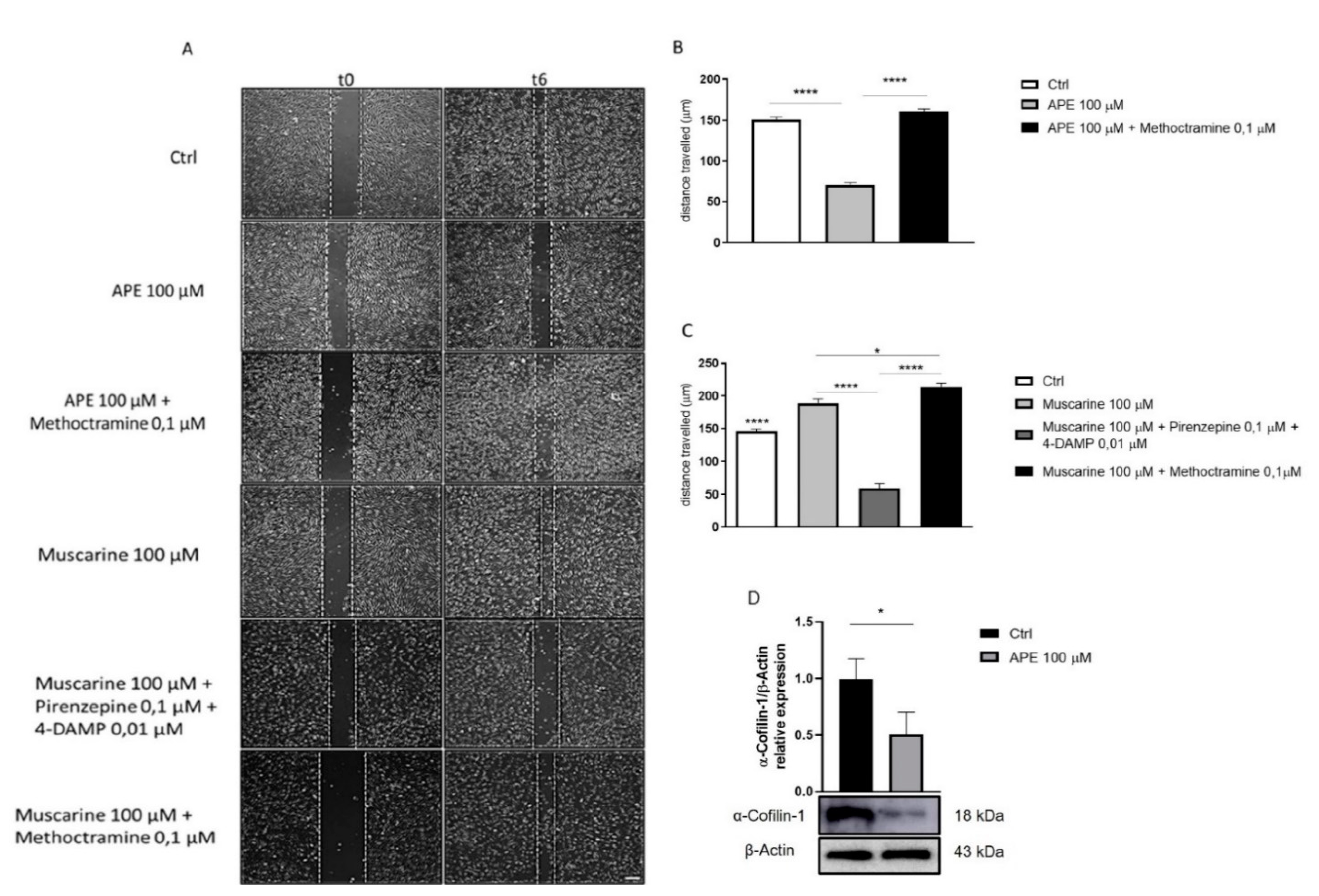{kind=link}
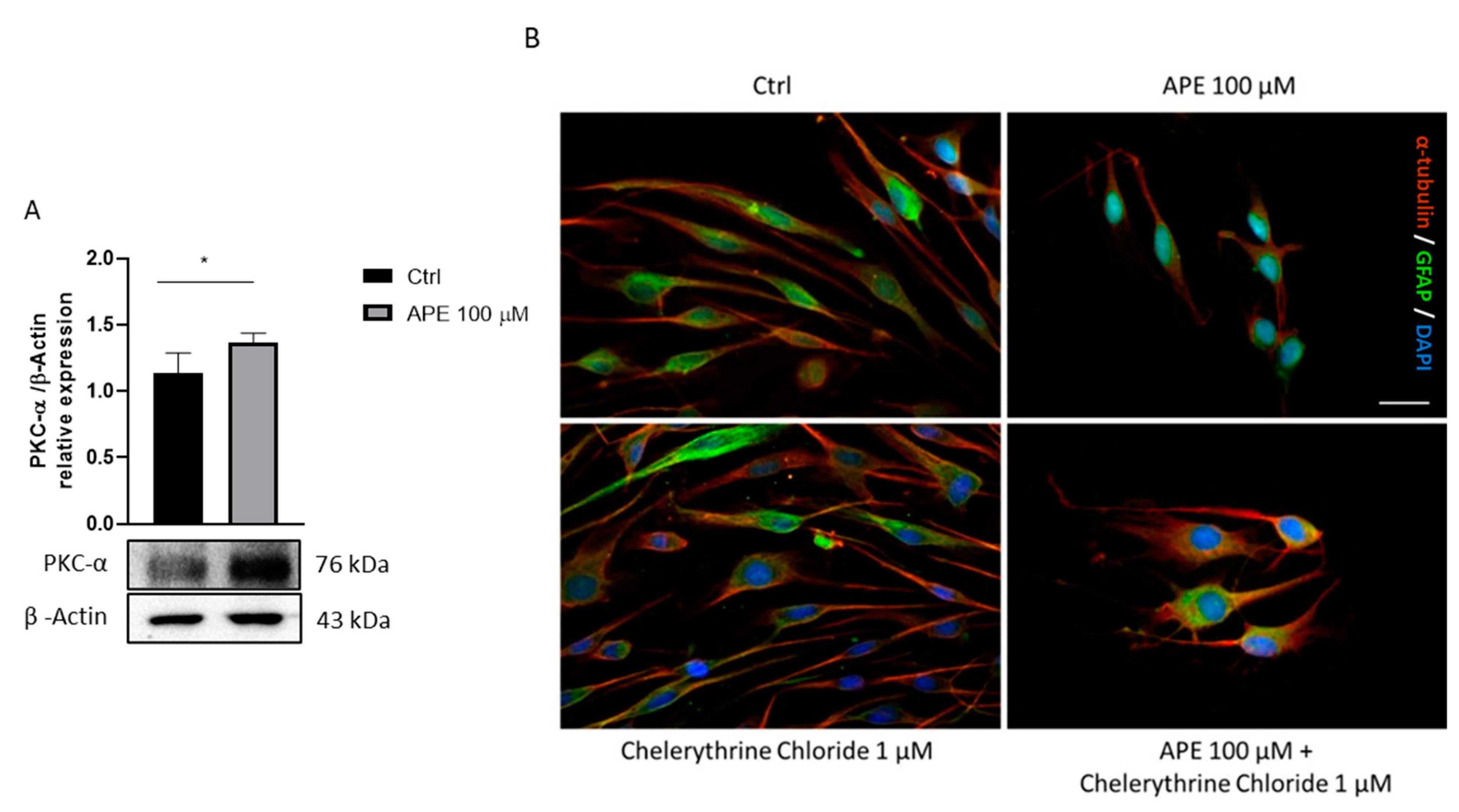{kind=link}
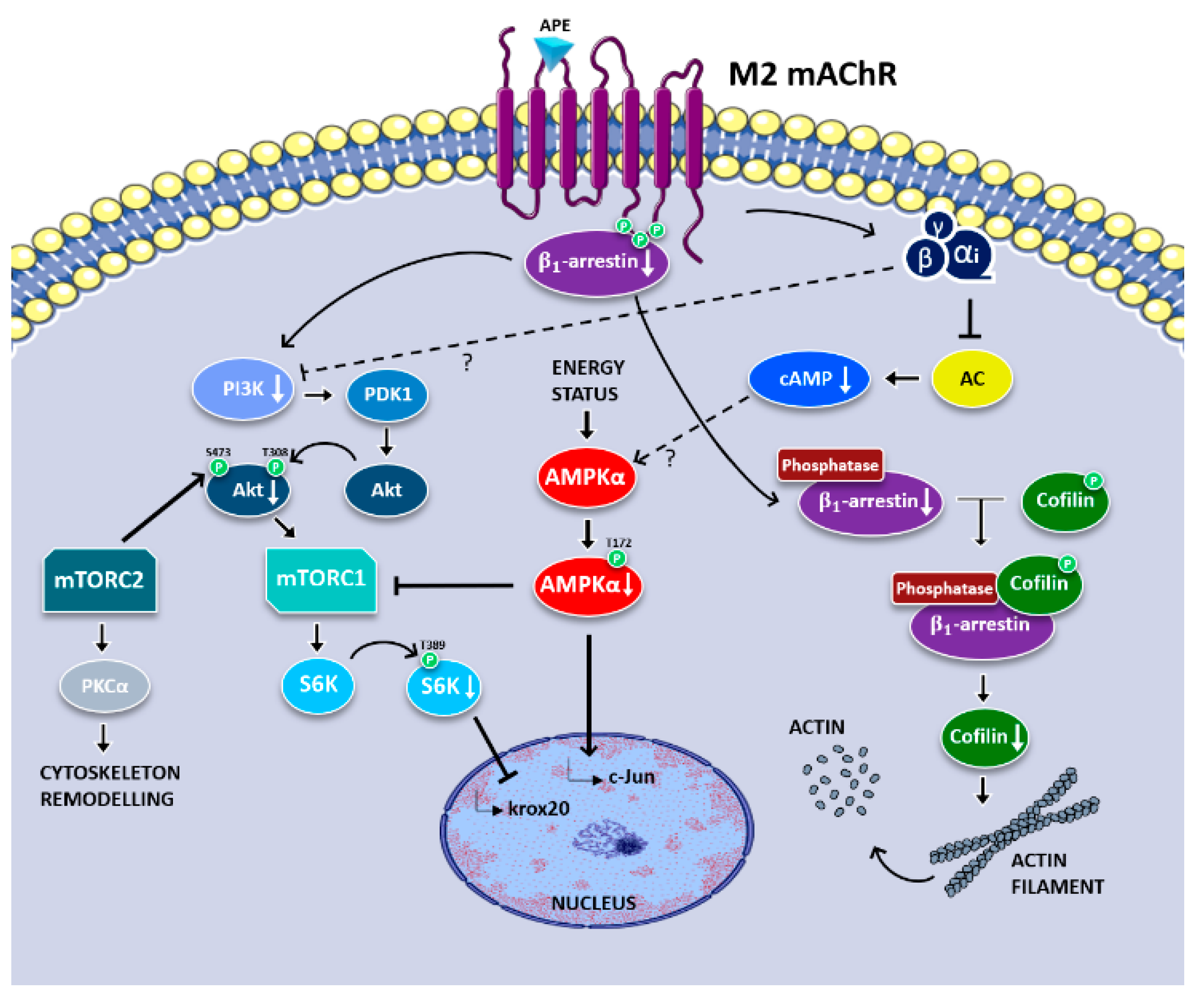{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Statements for Animal Use
2.2. Cell Cultures
2.3. Pharmacological Treatment
2.4. Western Blot Analysis
2.5. Immunocytochemistry
2.6. Wound Healing
2.7. Statistical Analysis
3. Results
3.1. Analysis of Signaling Pathways Downstream M2 Receptor Activation
3.2. M2 Receptor Activation Results in SC Migration Arrest
3.3. M2 Receptor Activation Induces Morphological Change in SCs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Jessen, K.R.; Mirsky, R.; Lloyd, A.C. Schwann cells: Development and role in nerve repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a020487. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Mirsky, R. The Repair Schwann cell and its function in regenerating nerves: Repair Schwann cell and its function in regenerating nerves. J. Physiol. 2016, 594, 3521–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessen, K.R.; Mirsky, R. The success and failure of the Schwann cell response to nerve injury. Front. Cell. Neurosci. 2019, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T. The role of neurotrophic factors in nerve regeneration. Neurosurg. Focus 2009, 26, E3. [Google Scholar] [CrossRef] [PubMed]
- Augusti-Tocco, G.; Biagioni, S.; Tata, A.M. Acetylcholine and regulation of gene expression in developing systems. J. Mol. Neurosci. 2006, 30, 45–48. [Google Scholar] [CrossRef]
- Magnaghi, V.; Procacci, P.; Tata, A.M. Chapter 15 novel pharmacological approaches to Schwann cells as neuroprotective agents for peripheral nerve regeneration. In International Review of Neurobiology; Elsevier: Amsterdam, The Netherlands, 2009; Volume 87, pp. 295–315. ISBN 978-0-12-375084-6. [Google Scholar]
- Fields, R.D. Release of neurotransmitters from Glia. Neuron Glia Biol. 2010, 6, 137–139. [Google Scholar] [CrossRef] [Green Version]
- Loreti, S.; Vilaró, M.T.; Visentin, S.; Rees, H.; Levey, A.I.; Tata, A.M. Rat Schwann cells express M1–M4 muscarinic receptor subtypes. J. Neurosci. Res. 2006, 84, 97–105. [Google Scholar] [CrossRef]
- Loreti, S.; Ricordy, R.; De Stefano, M.E.; Augusti-Tocco, G.; Tata, A.M. Acetylcholine inhibits cell cycle progression in rat Schwann cells by activation of the M2 receptor subtype. Neuron Glia Biol. 2007, 3, 269–279. [Google Scholar] [CrossRef]
- Uggenti, C.; De Stefano, M.E.; Costantino, M.; Loreti, S.; Pisano, A.; Avallone, B.; Talora, C.; Magnaghi, V.; Tata, A.M. M2 muscarinic receptor activation regulates schwann cell differentiation and Myelin organization: ACh modulation of Schwann cell development. Dev. Neurobiol. 2014, 74, 676–691. [Google Scholar] [CrossRef]
- Piovesana, R.; Faroni, A.; Tata, A.M.; Reid, A.J. Functional characterization of muscarinic receptors in human Schwann cells. Int. J. Mol. Sci. 2020, 21, 6666. [Google Scholar] [CrossRef]
- Iacovelli, J.; Lopera, J.; Bott, M.; Baldwin, E.; Khaled, A.; Uddin, N.; Fernandez-Valle, C. Serum and forskolin cooperate to promote G1 progression in schwann cells by differentially regulating cyclin D1, cyclin E1, and P27 Kip expression. Glia 2007, 55, 1638–1647. [Google Scholar] [CrossRef] [PubMed]
- Rahmatullah, M.; Schroering, A.; Rothblum, K.; Stahl, R.C.; Urban, B.; Carey, D.J. Synergistic regulation of Schwann cell proliferation by Heregulin and Forskolin. Mol. Cell. Biol. 1998, 18, 6245–6252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopal, S.; Shenoy, S.K. GPCR desensitization: Acute and prolonged phases. Cell. Signal. 2018, 41, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Bünemann, M.; Hosey, M.M. G-protein coupled receptor kinases as modulators of G-protein signalling. J. Physiol. 1999, 517, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor–β-arrestin complex in a lipid nanodisc. Nature 2020, 579, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Daaka, Y.; Della Rocca, G.J.; Lefkowitz, R.J. G Protein-coupled receptors mediate two functionally distinct pathways of tyrosine phosphorylation in rat 1a fibroblasts. J. Biol. Chem. 1997, 272, 31648–31656. [Google Scholar] [CrossRef] [Green Version]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. β-arrestins and cell signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef] [Green Version]
- Figlia, G.; Norrmén, C.; Pereira, J.A.; Gerber, D.; Suter, U. Dual function of the PI3K-Akt-MTORC1 axis in myelination of the peripheral nervous system. eLife 2017, 6, e29241. [Google Scholar] [CrossRef]
- Maurel, P.; Salzer, J.L. Axonal Regulation of Schwann cell proliferation and survival and the initial events of myelination requires PI 3-kinase activity. J. Neurosci. 2000, 20, 4635–4645. [Google Scholar] [CrossRef] [Green Version]
- Piovesana, R.; Faroni, A.; Magnaghi, V.; Reid, A.J.; Tata, A.M. M2 receptors activation modulates cell growth, migration and differentiation of rat Schwann-like adipose-derived stem cells. Cell Death Discov. 2019, 5, 92. [Google Scholar] [CrossRef]
- Alessandrini, F.; Cristofaro, I.; Di Bari, M.; Zasso, J.; Conti, L.; Tata, A.M. The activation of M2 muscarinic receptor inhibits cell growth and survival in human glioblastoma cancer stem cells. Int. Immunopharmacol. 2015, 29, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M.; Fabbiano, C.; Di Bari, M.; Conte, C.; Castigli, E.; Sciaccaluga, M.; Ponti, D.; Ruggieri, P.; Raco, A.; Ricordy, R.; et al. M2 receptor activation inhibits cell cycle progression and survival in human glioblastoma cells. J. Cell. Mol. Med. 2013, 17, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Lucianò, A.M.; Perciballi, E.; Fiore, M.; Del Bufalo, D.; Tata, A.M. The combination of the M2 muscarinic receptor agonist and chemotherapy affects drug resistance in neuroblastoma cells. Int. J. Mol. Sci. 2020, 21, 8433. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, M.; Tombolillo, V.; Alessandrini, F.; Guerriero, C.; Fiore, M.; Asteriti, I.A.; Castigli, E.; Sciaccaluga, M.; Guarguaglini, G.; Degrassi, F.; et al. M2 Muscarinic receptor activation impairs mitotic progression and bipolar mitotic spindle formation in human glioblastoma cell lines. Cells 2021, 10, 1727. [Google Scholar] [CrossRef] [PubMed]
- Tata, A.M.; Biagioni, S.; Ricci, A.; Amenta, F.; Augusti-Tocco, G. Muscarinic cholinergic receptors in dorsal root ganglia of chick embryo: A radioligand binding and immunocytochemical study. Neurosci. Lett. 1995, 189, 139–142. [Google Scholar] [CrossRef]
- Salani, M.; Anelli, T.; Tocco, G.A.; Lucarini, E.; Mozzetta, C.; Poiana, G.; Tata, A.M.; Biagioni, S. Acetylcholine-induced neuronal differentiation: Muscarinic receptor activation regulates EGR-1 and REST expression in neuroblastoma cells. J. Neurochem. 2009, 108, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Ghashghaeinia, M.; Koralkova, P.; Giustarini, D.; Mojzikova, R.; Fehrenbacher, B.; Dreischer, P.; Schaller, M.; Mrowietz, U.; Martínez-Ruiz, A.; Wieder, T.; et al. The Specific PKC-α inhibitor chelerythrine blunts costunolide-induced eryptosis. Apoptosis 2020, 25, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Marshall, F.H. Visualizing GPCR ‘Megaplexes’ which enable sustained intracellular signaling. Trends Biochem. Sci. 2016, 41, 985–986. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, A.R.B.; Plouffe, B.; Cahill, T.J.; Shukla, A.K.; Tarrasch, J.T.; Dosey, A.M.; Kahsai, A.W.; Strachan, R.T.; Pani, B.; Mahoney, J.P.; et al. GPCR-G protein-β-arrestin super-complex mediates sustained G protein signaling. Cell 2016, 166, 907–919. [Google Scholar] [CrossRef] [Green Version]
- Efeyan, A.; Sabatini, D.M. MTOR and cancer: Many loops in one pathway. Curr. Opin. Cell Biol. 2010, 22, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Peng, S.; Zhao, Y.; Zhao, T.; Wang, M.; Luo, L.; Yang, Y.; Sun, C. AMPK Negatively regulates peripheral myelination via activation of C-Jun. Mol. Neurobiol. 2017, 54, 3554–3564. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Corum, L.; Meng, Q.; Blenis, J.; Zheng, J.Z.; Shi, X.; Flynn, D.C.; Jiang, B.-H. PI3K induced actin filament remodeling through Akt and P70S6K1: Implication of essential role in cell migration. Am. J. Physiol.-Cell Physiol. 2004, 286, C153–C163. [Google Scholar] [CrossRef] [PubMed]
- McGovern, K.W.; DeFea, K.A. Molecular mechanisms underlying beta-arrestin-dependent chemotaxis and actin-cytoskeletal reorganization. In Arrestins—Pharmacology and Therapeutic Potential; Gurevich, V.V., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 219, pp. 341–359. ISBN 978-3-642-41198-4. [Google Scholar]
- Singh, R.K.; Kumar, S.; Gautam, P.K.; Tomar, M.S.; Verma, P.K.; Singh, S.P.; Kumar, S.; Acharya, A. Protein Kinase C-α and the regulation of diverse cell responses. Biomol. Concepts 2017, 8, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damato, M.; Cardon, T.; Wisztorski, M.; Fournier, I.; Pieragostino, D.; Cicalini, I.; Salzet, M.; Vergara, D.; Maffia, M. Protein Kinase C activation drives a differentiation program in an oligodendroglial precursor model through the modulation of specific biological networks. Int. J. Mol. Sci. 2021, 22, 5245. [Google Scholar] [CrossRef]
- De Angelis, F.; Bernardo, A.; Magnaghi, V.; Minghetti, L.; Tata, A.M. Muscarinic receptor subtypes as potential targets to modulate oligodendrocyte progenitor survival, proliferation, and differentiation. Dev. Neurobiol. 2012, 72, 713–728. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. Negative regulation of myelination: Relevance for development, injury, and demyelinating disease. Glia 2008, 56, 1552–1565. [Google Scholar] [CrossRef]
- Stolt, C.C.; Wegner, M. Schwann cells and their transcriptional network: Evolution of key regulators of peripheral myelination. Brain Res. 2016, 1641, 101–110. [Google Scholar] [CrossRef]
- Ryu, E.J.; Wang, J.Y.T.; Le, N.; Baloh, R.H.; Gustin, J.A.; Schmidt, R.E.; Milbrandt, J. Misexpression of Pou3f1 results in peripheral nerve hypomyelination and axonal loss. J. Neurosci. 2007, 27, 11552–11559. [Google Scholar] [CrossRef] [Green Version]
- Bremer, M.; Fröb, F.; Kichko, T.; Reeh, P.; Tamm, E.R.; Suter, U.; Wegner, M. Sox10 is required for Schwann-cell homeostasis and myelin maintenance in the adult peripheral nerve. Glia 2011, 59, 1022–1032. [Google Scholar] [CrossRef]
- Schuh, C.M.A.P.; Sandoval-Castellanos, A.M.; De Gregorio, C.; Contreras-Kallens, P.; Haycock, J.W. The role of Schwann cells in peripheral nerve function, injury, and repair. In Cell Engineering and Regeneration; Gimble, J.M., Marolt Presen, D., Oreffo, R., Redl, H., Wolbank, S., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 1–22. ISBN 978-3-319-37076-7. [Google Scholar]
- Norrmén, C.; Figlia, G.; Lebrun-Julien, F.; Pereira, J.A.; Trötzmüller, M.; Köfeler, H.C.; Rantanen, V.; Wessig, C.; van Deijk, A.-L.F.; Smit, A.B.; et al. MTORC1 controls PNS myelination along the MTORC1-RXRγ-SREBP-lipid biosynthesis axis in Schwann cells. Cell Rep. 2014, 9, 646–660. [Google Scholar] [CrossRef] [Green Version]
- Figlia, G.; Gerber, D.; Suter, U. Myelination and MTOR. Glia 2018, 66, 693–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norrmén, C.; Suter, U. Akt/MTOR signalling in myelination. Biochem. Soc. Trans. 2013, 41, 944–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, T.; Yamamoto, S.; Nakamura, K.; Tanaka, S. Signaling axis in Schwann cell proliferation and differentiation. Mol. Neurobiol. 2006, 33, 51–61. [Google Scholar] [CrossRef]
- Ma, L.; Pei, G. β-arrestin signaling and regulation of transcription. J. Cell Sci. 2007, 120, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luttrell, L.M.; Lefkowitz, R.J. The Role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 2002, 115, 455–465. [Google Scholar] [CrossRef] [PubMed]
- van Gastel, J.; Hendrickx, J.O.; Leysen, H.; Santos-Otte, P.; Luttrell, L.M.; Martin, B.; Maudsley, S. β-arrestin based receptor signaling paradigms: Potential therapeutic targets for complex age-related disorders. Front. Pharmacol. 2018, 9, 1369. [Google Scholar] [CrossRef] [PubMed]
- Santos-Otte, P.; Leysen, H.; van Gastel, J.; Hendrickx, J.O.; Martin, B.; Maudsley, S. G Protein-coupled receptor systems and their role in cellular senescence. Comput. Struct. Biotechnol. J. 2019, 17, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Jean-Charles, P.-Y.; Kaur, S.; Shenoy, S.K. G protein–coupled receptor signaling through β-arrestin–dependent mechanisms. J. Cardiovasc. Pharmacol. 2017, 70, 142–158. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Cotter, L.; Ozcelik, M.; Jacob, C.; Pereira, J.A.; Locher, V.; Baumann, R.; Relvas, J.B.; Suter, U.; Tricaud, N. Dlg1-PTEN interaction regulates myelin thickness to prevent damaging peripheral nerve overmyelination. Science 2010, 328, 1415–1418. [Google Scholar] [CrossRef]
- Beirowski, B.; Wong, K.M.; Babetto, E.; Milbrandt, J. MTORC1 promotes proliferation of immature schwann cells and myelin growth of differentiated Schwann cells. Proc. Natl. Acad. Sci. USA 2017, 114, E4261–E4270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boerboom, A.; Dion, V.; Chariot, A.; Franzen, R. Molecular mechanisms involved in Schwann cell plasticity. Front. Mol. Neurosci. 2017, 10, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunge, R.P. Expanding roles for the Schwann cell: Ensheathment, myelination, trophism and regeneration. Curr. Opin. Neurobiol. 1993, 3, 805–809. [Google Scholar] [CrossRef]
- Clark, M.B.; Bunge, M.B. Cultured Schwann cells assemble normal-appearing basal lamina only when they ensheathe axons. Dev. Biol. 1989, 133, 393–404. [Google Scholar] [CrossRef]
- Yamauchi, J.; Chan, J.R.; Shooter, E.M. Neurotrophins regulate Schwann cell migration by activating divergent signaling pathways dependent on Rho GTPases. Proc. Natl. Acad. Sci. USA 2004, 101, 8774–8779. [Google Scholar] [CrossRef] [Green Version]
- Kakinuma, N.; Roy, B.C.; Zhu, Y.; Wang, Y.; Kiyama, R. Kank regulates RhoA-dependent formation of actin stress fibers and cell migration via 14-3-3 in PI3K–Akt signaling. J. Cell Biol. 2008, 181, 537–549. [Google Scholar] [CrossRef]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [Green Version]
- Ma, A.D.; Metjian, A.; Bagrodia, S.; Taylor, S.; Abrams, C.S. Cytoskeletal reorganization by G protein-coupled receptors is dependent on phosphoinositide 3-kinase γ, a rac guanosine exchange factor, and Rac. Mol. Cell. Biol. 1998, 18, 4744–4751. [Google Scholar] [CrossRef] [Green Version]
- Min, J.; DeFea, K. β-arrestin-dependent actin reorganization: Bringing the right players together at the leading edge. Mol. Pharmacol. 2011, 80, 760–768. [Google Scholar] [CrossRef]
- Zoudilova, M.; Min, J.; Richards, H.L.; Carter, D.; Huang, T.; DeFea, K.A. β-arrestins scaffold cofilin with chronophin to direct localized actin filament severing and membrane protrusions downstream of protease-activated receptor-2. J. Biol. Chem. 2010, 285, 14318–14329. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Huang, J.; Yuan, Z.; Feng, S.; Xie, Y.; Ma, W. Protein kinase C inhibitor chelerythrine selectively inhibits proliferation of triple-negative breast cancer cells. Sci. Rep. 2017, 7, 2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbert, J.M.; Augereau, J.M.; Gleye, J.; Maffrand, J.P. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 1990, 172, 993–999. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botticelli, E.; Salazar Intriago, M.S.; Piovesana, R.; Tata, A.M. Analysis of Signal Transduction Pathways Downstream M2 Receptor Activation: Effects on Schwann Cell Migration and Morphology. Life 2022, 12, 211. https://doi.org/10.3390/life12020211
Botticelli E, Salazar Intriago MS, Piovesana R, Tata AM. Analysis of Signal Transduction Pathways Downstream M2 Receptor Activation: Effects on Schwann Cell Migration and Morphology. Life. 2022; 12(2):211. https://doi.org/10.3390/life12020211
Chicago/Turabian StyleBotticelli, Elisabetta, Michael Sebastian Salazar Intriago, Roberta Piovesana, and Ada Maria Tata. 2022. "Analysis of Signal Transduction Pathways Downstream M2 Receptor Activation: Effects on Schwann Cell Migration and Morphology" Life 12, no. 2: 211. https://doi.org/10.3390/life12020211
APA StyleBotticelli, E., Salazar Intriago, M. S., Piovesana, R., & Tata, A. M. (2022). Analysis of Signal Transduction Pathways Downstream M2 Receptor Activation: Effects on Schwann Cell Migration and Morphology. Life, 12(2), 211. https://doi.org/10.3390/life12020211