
Comparison of Auxenochlorella protothecoides and Chlorella spp. Chloroplast Genomes: Evidence for Endosymbiosis and Horizontal Virus-like Gene Transfer



Abstract
:1. Introduction
2. Materials and Methods
2.1. Auxenochlorella protothecoides UTEX 25 Culture and DNA Isolation
2.2. Auxenochlorella protothecoides UTEX 25 Chloroplast Genome Sequencing and Annotation
2.3. Chloroplast Genome Sequence Alignment
2.4. Intron and Repeat Element Analysis
2.5. Bacterial and Viral Sequence Search
2.6. Phylogenetic Analyses
2.7. Analysis of Four Predicted Virus-like Transcripts
3. Results
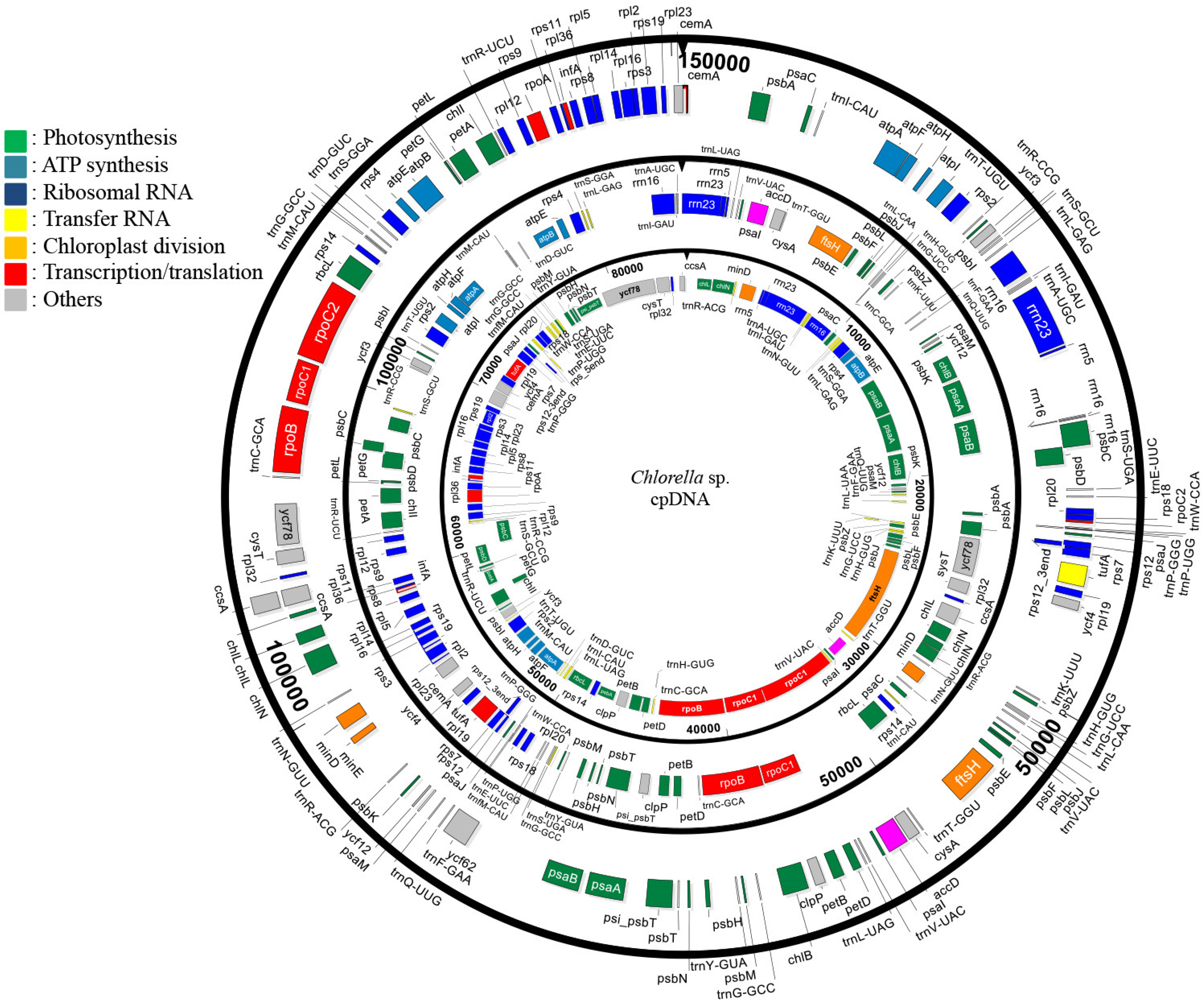
3.1. General Characteristics
3.2. Phylogenetic Analysis of Trebouxiophyceae Chloroplast Genomes
3.3. Plastid-Encoded RNA Polymerase
3.4. Chloroplast Division
3.5. Chlorophyll Synthesis
3.6. Chloroplast Introns
3.7. Transposable Element (TE) and Repeated Sequence Analysis
3.8. Endosymbiotic Cyanobacteria and Viral Signatures
4. Discussion
4.1. Genome Comparison
4.2. Evolutionary Implications
4.3. Future Biotechnological Impact
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wells, M.L.; Potin, P.; Craigie, J.S.; Raven, J.A.; Merchant, S.S.; Helliwell, K.E.; Smith, A.G.; Camire, M.E.; Brawley, S.H. Algae as nutritional and functional food sources: Revisiting our understanding. J. Appl. Phycol. 2017, 29, 949–982. [Google Scholar] [CrossRef] [PubMed]
- Thiyagarasaiyar, K.; Goh, B.H.; Jeon, Y.J.; Yow, Y.Y. Algae Metabolites in Cosmeceutical: An Overview of Current Applications and Challenges. Mar. Drugs 2020, 18, 323. [Google Scholar] [CrossRef] [PubMed]
- Harwood, J.L. Algae: Critical sources of very long-chain polyunsaturated fatty acids. Biomolecules 2019, 9, 708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannan, M.A.; Dash, R.; Haque, M.N.; Mohibbullah, M.; Sohag, A.; Rahman, M.A.; Uddin, M.J.; Alam, M.; Moon, I.S. Neuroprotective potentials of marine algae and their bioactive metabolites: Pharmacological insights and therapeutic advances. Mar. Drugs 2020, 18, 347. [Google Scholar] [CrossRef]
- Xiao, Y.; He, X.; Ma, Q.; Lu, Y.; Bai, F.; Dai, J.; Wu, Q. Photosynthetic Accumulation of Lutein in Auxenochlorella protothecoides after Heterotrophic Growth. Mar. Drugs 2018, 16, 283. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.I.; Shin, J.H.; Kim, J.D. The promising future of microalgae: Current status, challenges, and optimization of a sustainable and renewable industry for biofuels, feed, and other products. Microb. Cell Fact. 2018, 17, 36. [Google Scholar] [CrossRef]
- Khalili, A.; Najafpour, G.D.; Amini, G.; Samkhaniyani, F. Influence of nutrients and LED light intensities on biomass production of microalgae Chlorella vulgaris. Biotech. Bioprocess. Eng. 2015, 20, 284–290. [Google Scholar] [CrossRef]
- Kula, M.; Rys, M.; Skoczowski, A. Far-red light (720 or 740 nm) improves growth and changes the chemical composition of Chlorella vulgaris. Eng. Life Sci. 2014, 14, 651–657. [Google Scholar] [CrossRef]
- Singh, S.P.; Singh, P. Effect of CO2 concentration on algal growth: A review. Renew. Sustain. Energy Rev. 2014, 38, 172–179. [Google Scholar] [CrossRef]
- Rismani-Yazdi, H.; Hampel, K.H.; Lane, C.D.; Kessler, B.A.; White, N.M.; Moats, K.M.; Thomas Allnutt, F.C. High-productivity lipid production using mixed trophic state cultivation of Auxenochlorella (Chlorella) protothecoides. Bioprocess. Biosyst. Eng. 2015, 38, 639–650. [Google Scholar] [CrossRef]
- Zhang, J.H.; Hao, Q.; Bai, L.L.; Xu, J.; Yin, W.; Song, L.; Xu, L.; Guo, X.; Fan, C.; Chen, Y.; et al. Overexpression of the soybean transcription factor GmDof4 significantly enhances the lipid content of Chlorella ellipsoidea. Biotechnol. Biofuels 2014, 7, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.H.; Kyndt, J.; Chougule, K.; Park, J.-J.; Brown, J.K. Low-phosphate-selected Auxenochlorella protothecoides redirects phosphate to essential pathways while producing more biomass. PLoS ONE 2018, 13, e0198953. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Krienitz, L.; Proschold, T. Taxonomic reassessment of the genus Chlorella (Trebouxiophyceae) using molecular signatures (barcodes), including description of seven new species. Fottea 2011, 11, 293–312. [Google Scholar] [CrossRef] [Green Version]
- Leliaert, F.; Smith, D.R.; Moreau, H.; Herron, M.D.; Verbruggen, H.; Delwiche, C.F.; De Clerck, O. Phylogeny and molecular evolution of the green algae. Crit. Rev. Plant Sci. 2012, 31, 1–46. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Fang, L.; Zhang, Z.; Chang, X.; Penny, D.; Zhong, B. Chloroplast Phylogenomic Inference of Green Algae Relationships. Sci. Rep. 2016, 6, 20528. [Google Scholar] [CrossRef] [Green Version]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [Green Version]
- Brouard, J.-S.; Otis, C.; Lemieux, C.; Turmel, M. The exceptionally large chloroplast genome of the green alga Floydiella terrestris illuminates the evolutionary history of the Chlorophyceae. Genome Biol. Evol. 2010, 2, 240–256. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zheng, Y. Dynamic evolution and phylogenomic analysis of the chloroplast genome in Schisandraceae. Sci. Rep. 2018, 8, 9285. [Google Scholar] [CrossRef]
- Serrato-Capuchina, A.; Matute, D.R. The role of transposable elements in speciation. Genes 2018, 9, 254. [Google Scholar] [CrossRef] [Green Version]
- Whatley, J.M. The Endosymbiotic Origin of Chloroplasts. In International Review of Cytology; Jeon, K.W., Jarvik, J., Eds.; Academic Press: Cambridge, MA, USA, 1993; pp. 259–299. [Google Scholar]
- Turmel, M.; Otis, C.; Lemieux, C. The complete chloroplast DNA sequence of the green alga Nephroselmis olivacea: Insights into the architecture of ancestral chloroplast genomes. Proc. Nat. Acad. Sci. USA 1999, 96, 10248–10253. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Yoon, H.S.; Bhattacharya, D. Algal endosymbionts as vectors of horizontal gene transfer in photosynthetic eukaryotes. Front. Plant Sci. 2013, 4, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaroque, N.; Boland, W. The genome of the brown alga Ectocarpus siliculosus contains a series of viral DNA pieces, suggesting an ancient association with large dsDNA viruses. BMC Evol. Biol. 2008, 8, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leliaert, F.; Lopez-Bautista, J.M. The chloroplast genomes of Bryopsis plumosa and Tydemania expeditiones (Bryopsidales, Chlorophyta): Compact genomes and genes of bacterial origin. BMC Genom. 2015, 16, 204. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.R. Haematococcus lacustris: The makings of a giant-sized chloroplast genome. AoB Plants 2018, 10, ply058. [Google Scholar] [CrossRef]
- Turmel, M.; Otis, C.; Lemieux, C. Divergent copies of the large inverted repeat in the chloroplast genomes of ulvophycean green algae. Sci. Rep. 2017, 7, 994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulieu, J.M.; Leitch, I.J.; Patel, S.; Pendharkar, A.; Knight, C.A. Genome size is a strong predictor of cell size and stomatal density in angiosperms. New Phytol. 2008, 179, 975–986. [Google Scholar] [CrossRef] [Green Version]
- Cavalier-Smith, T. Economy, speed and size matter: Evolutionary forces driving nuclear genome miniaturization and expansion. Ann. Bot. 2005, 95, 147–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, E.L.; Wallau, G.L.; Rangel, D.L.; Machado, L.C.; Pereira, A.B.; Victoria, F.C.; Boldo, J.T.; Pinto, P.M. Phylogenetic positioning of the Antarctic alga Prasiola crispa (Trebouxiophyceae) using organellar genomes and their structural analysis. J. Phycol. 2017, 53, 908–915. [Google Scholar] [CrossRef]
- Cremen, M.C.M.; Leliaert, F.; West, J.; Lam, D.W.; Shimada, S.; Lopez-Bautista, J.M.; Verbruggen, H. Reassessment of the classification of Bryopsidales (Chlorophyta) based on chloroplast phylogenomic analyses. Mol. Phylogenetics Evol. 2019, 130, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Hovde, B.T.; Hanschen, E.R.; Steadman, C.R.; Lo, C.-C.; Kunde, Y.; Davenport, K.; Daligault, H.; Msanne, J.; Canny, S.; Eyun, S.; et al. Genomic characterization reveals significant divergence within Chlorella sorokiniana (Chlorellales, Trebouxiophyceae). Algal Res. 2018, 35, 449–461. [Google Scholar] [CrossRef]
- Satjarak, A.; Graham, L.E. Comparative DNA sequence analyses of Pyramimonas parkeae (Prasinophyceae) chloroplast genomes. J. Phycol. 2017, 53, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Wang, L.; Zhou, L.; He, P.; Jiao, B. Complete chloroplast genome of green tide algae Ulva flexuosa (Ulvophyceae, Chlorophyta) with comparative analysis. PLoS ONE 2017, 12, e0184196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orsini, M.; Costelli, C.; Malavasi, V.; Cusano, R.; Concas, A.; Angius, A.; Cao, G. Complete sequence and characterization of mitochondrial and chloroplast genome of Chlorella variabilis NC64A. Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 2016, 27, 3128–3130. [Google Scholar] [CrossRef] [PubMed]
- Han, C.S.; Chain, P. Finishing repeat regions automatically with Dupfinisher. In Proceedings of the 2006 International Conference on Bioinformatics & Computational Biology, Las Vegas, NV, USA, 26–29 June 2006; pp. 141–146. [Google Scholar]
- Gordon, D.; Abajian, C.; Green, P. Consed: A graphical tool for sequence finishing. Genome Res. 1998, 8, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lu, C.; Wu, Q.J.; Wang, Y.; Sun, Z.T.; Deng, J.C.; Zhang, Y. GISSD: Group I intron sequence and structure database. Nucleic Acids Res. 2008, 36, D31–D37. [Google Scholar] [CrossRef]
- Dai, L.; Toor, N.; Olson, R.; Keeping, A.; Zimmerly, S. Database for mobile group II introns. Nucleic Acids Res. 2003, 31, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.; Hubley, R.; Green, P. RepeatMasker Open-3.0 (1996–2010). Available online: http://www.repeatmasker.org (accessed on 10 February 2022).
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Birky, C.W., Jr. Relaxed cellular controls and organelle heredity. Science 1983, 222, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemieux, C.; Otis, C.; Turmel, M. Chloroplast phylogenomic analysis resolves deep-level relationships within the green algal class Trebouxiophyceae. BMC Ecol. Evol. 2014, 14, 211. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Wang, Y.; Murakami, T.; Shen, Y.; Gong, J.; Jiang, H.; Smith, D.R.; Pombert, J.-F.; Dai, J.; Wu, Q. Auxenochlorella protothecoides and Prototheca wickerhamii plastid genome sequences give insight into the origins of non-photosynthetic algae. Sci. Rep. 2015, 5, 14465. [Google Scholar] [CrossRef] [Green Version]
- Higgins, B.T.; Nobles, D.; Ma, Y.; Wikoff, W.R.; Kind, T.; Fiehn, O.; Brand, J.; VanderGheynst, J.S. Informatics for improved algal taxonomic classification and research: A case study of UTEX 2341. Algal Res. 2015, 12, 545–549. [Google Scholar] [CrossRef] [Green Version]
- Serino, G.; Maliga, P. RNA polymerase subunits encoded by the plastid rpo genes are not shared with the nucleus-encoded plastid enzyme. Plant Physiol. 1998, 117, 1165–1170. [Google Scholar] [CrossRef] [Green Version]
- Harry, E.; Monahan, L.; Thompson, L. Bacterial cell division: The mechanism and its precison. Int. Rev. Cytol. 2006, 253, 27–94. [Google Scholar] [PubMed]
- Margolin, W. Bacterial cell division: A moving MinE sweeper boggles the MinD. Curr. Biol. 2001, 11, R395–R398. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.W.; Errington, J. Bacterial cell division: Assembly, maintenance and disassembly of the Z ring. Nat. Rev. Microbiol. 2009, 7, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Park, K.T.; Wu, W.; Battaile, K.P.; Lovell, S.; Holyoak, T.; Lutkenhaus, J. The Min oscillator uses MinD-dependent conformational changes in MinE to spatially regulate cytokinesis. Cell 2011, 146, 396–407. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, R.; Rajeswari, H.; Ajitkumar, P. Analysis of degradation of bacterial cell division protein FtsZ by the ATP-dependent zinc-metalloprotease FtsH in vitro. Microbiol. Res. 2008, 163, 21–30. [Google Scholar] [CrossRef]
- Jensen, P.E.; Reid, J.D.; Hunter, C.N. Modification of cysteine residues in the ChlI and ChlH subunits of magnesium chelatase results in enzyme inactivation. Biochem. J. 2000, 352, 435–441. [Google Scholar] [CrossRef]
- Jensen, P.E.; Gibson, L.C.; Henningsen, K.W.; Hunter, C.N. Expression of the chlI, chlD, and chlH genes from the Cyanobacterium Synechocystis PCC6803 in Escherichia coli and demonstration that the three cognate proteins are required for magnesium-protoporphyrin chelatase activity. J. Biol. Chem. 1996, 271, 16662–16667. [Google Scholar] [CrossRef] [Green Version]
- Blanc, G.; Duncan, G.; Agarkova, I.; Borodovsky, M.; Gurnon, J.; Kuo, A.; Lindquist, E.; Lucas, S.; Pangilinan, J.; Polle, J.; et al. The Chlorella variabilis NC64A genome reveals adaptation to photosymbiosis, coevolution with viruses, and cryptic sex. Plant Cell 2010, 22, 2943–2955. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Mao, Y.; Kong, F.; Li, G.; Ma, F.; Zhang, B.; Sun, P.; Bi, G.; Zhang, F.; Xue, H. Complete sequence and analysis of plastid genomes of two economically important red algae: Pyropia haitanensis and Pyropia yezoensis. PLoS ONE 2013, 8, e65902. [Google Scholar] [CrossRef] [Green Version]
- Cahoon, A.B.; Timko, M.P. yellow-in-the-dark mutants of Chlamydomonas lack the CHLL subunit of light-independent protochlorophyllide reductase. Plant Cell 2000, 12, 559–568. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Goldschmidt-Clermont, M.; Timko, M.P. Chloroplast-encoded chlB is required for light-independent protochlorophyllide reductase activity in Chlamydomonas reinhardtii. Plant Cell 1993, 5, 1817–1829. [Google Scholar] [PubMed] [Green Version]
- Saldanha, R.; Mohr, G.; Belfort, M.; Lambowitz, A.M. Group I and group II introns. FASEB J. 1993, 7, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Lambowitz, A.M.; Zimmerly, S. Mobile group II introns. Annu. Rev. Genet. 2004, 38, 1–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, W.J. Genetic instability and fragmentation of a stealth viral genome. Pathobiology 1996, 64, 9–17. [Google Scholar] [CrossRef]
- Martin, W.J. Bacteria-related sequences in a simian cytomegalovirus-derived stealth virus culture. Exp. Mol. Pathol. 1999, 66, 8–14. [Google Scholar] [CrossRef]
- Suzuki, S.; Endoh, R.; Manabe, R.I.; Ohkuma, M.; Hirakawa, Y. Multiple losses of photosynthesis and convergent reductive genome evolution in the colourless green algae. Prototheca Sci. Rep. 2018, 8, 940. [Google Scholar] [CrossRef] [Green Version]
- Steiner, S.; Schroter, Y.; Pfalz, J.; Pfannschmidt, T. Identification of essential subunits in the plastid-encoded RNA polymerase complex reveals building blocks for proper plastid development. Plant Physiol. 2011, 157, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Wang, Y.; Shen, Y.; Yan, D.; He, X.; Dai, J.; Wu, Q. Oil accumulation mechanisms of the oleaginous microalga Chlorella protothecoides revealed through its genome, transcriptomes, and proteomes. BMC Genom. 2014, 15, 582. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Nosenko, T.; Hackett, J.D.; Bhattacharya, D. Phylogenomic analysis identifies red algal genes of endosymbiotic origin in the chromalveolates. Mol. Biol. Evol. 2006, 23, 663–674. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, M.; Masuda, T.; Bando, T.; Yamagata, H.; Ohta, H.; Takamiya, K. Cloning and expression of the soybean chlH gene encoding a subunit of Mg-chelatase and localization of the Mg2+ concentration-dependent ChlH protein within the chloroplast. Plant Cell Physiol. 1998, 39, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Bowen, N.J.; Jordan, I.K. Transposable elements and the evolution of eukaryotic complexity. Curr. Issues Mol. Biol. 2002, 4, 65–76. [Google Scholar]
- Fedoroff, N.V. Transposable elements as a molecular evolutionary force. Ann. N. Y. Acad. Sci. 1999, 870, 251–264. [Google Scholar] [CrossRef]
- Smith, D.R.; Hamaji, T.; Olson, B.J.; Durand, P.M.; Ferris, P.; Michod, R.E.; Featherston, J.; Nozaki, H.; Keeling, P.J. Organelle genome complexity scales positively with organism size in volvocine green algae. Mol. Biol. Evol. 2013, 30, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Bento, M.C.; Canha, R.; Eira, C.; Vingada, J.; Nicolau, L.; Ferreira, M.; Domingo, M.; Tavares, L.; Duarte, A. Herpesvirus infection in marine mammals: A retrospective molecular survey of stranded cetaceans in the Portuguese coastline. Infect. Genet. Evol. 2019, 67, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Sharon, I.; Alperovitch, A.; Rohwer, F.; Haynes, M.; Glaser, F.; Atamna-Ismaeel, N.; Pinter, R.Y.; Partensky, F.; Koonin, E.V.; Wolf, Y.I.; et al. Photosystem I gene cassettes are present in marine virus genomes. Nature 2009, 461, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Larbig, K.D.; Christmann, A.; Johann, A.; Klockgether, J.; Hartsch, T.; Merkl, R.; Wiehlmann, L.; Fritz, H.J.; Tümmler, B. Gene islands integrated into tRNA(Gly) genes confer genome diversity on a Pseudomonas aeruginosa clone. J. Bacteriol. 2002, 184, 6665–6680. [Google Scholar] [CrossRef] [Green Version]
- Moreau, H.; Piganeau, G.; Desdevises, Y.; Cooke, R.; Derelle, E.; Grimsley, N. Marine prasinovirus genomes show low evolutionary divergence and acquisition of protein metabolism genes by horizontal gene transfer. J. Virol. 2010, 84, 12555–12563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeanniard, A.; Dunigan, D.D.; Gurnon, J.R.; Agarkova, I.V.; Kang, M.; Vitek, J.; Duncan, G.; McClung, O.W.; Larsen, M.; Claverie, J.-M.; et al. Towards defining the chloroviruses: A genomic journey through a genus of large DNA viruses. BMC Genom. 2013, 14, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li-Beisson, Y.; Thelen, J.J.; Fedosejevs, E.; Harwood, J.L. The lipid biochemistry of eukaryotic algae. Prog. Lipid Res. 2019, 74, 31–68. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Fan, C.; Chen, Y.; Hu, Z. The potential for microalgae as bioreactors to produce pharmaceuticals. Int. J. Mol. Sci. 2016, 17, 962. [Google Scholar] [CrossRef] [Green Version]
- Morales-Sánchez, D.; Kyndt, J.; Ogden, K.; Martinez, A. Toward an understanding of lipid and starch accumulation in microalgae: A proteomic study of Neochloris oleoabundans cultivated under N-limited heterotrophic conditions. Algal Res. 2016, 20, 22–34. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. protothecoides (GenBank Accession No. KC631634.1) | C. variabilis (GenBank Accession No. KJ718922.1) | C. vulgaris (GenBank Accession No. NC001865.1) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Number * | Length (bp) | Percentage (%) | Number * | Length (bp) | Percentage (%) | Number * | Length (bp) | Percentage (%) | |
| TE elements | - | - | - | - | - | - | - | - | - |
| Small RNA | 3 | 1892 | 2.23 | 2 | 1533 | 1.23 | 3 | 936 | 0.62 |
| Simple repeats | 3 | 156 | 0.18 | 1 | 34 | 0.02 | 27 | 958 | 0.63 |
| Low complexity | 135 | 10,129 | 11.9 | 45 | 1858 | 1.49 | 168 | 7725 | 5.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.-H.; Kyndt, J.A.; Brown, J.K. Comparison of Auxenochlorella protothecoides and Chlorella spp. Chloroplast Genomes: Evidence for Endosymbiosis and Horizontal Virus-like Gene Transfer. Life 2022, 12, 458. https://doi.org/10.3390/life12030458
Park S-H, Kyndt JA, Brown JK. Comparison of Auxenochlorella protothecoides and Chlorella spp. Chloroplast Genomes: Evidence for Endosymbiosis and Horizontal Virus-like Gene Transfer. Life. 2022; 12(3):458. https://doi.org/10.3390/life12030458
Chicago/Turabian StylePark, Sang-Hyuck, John A. Kyndt, and Judith K. Brown. 2022. "Comparison of Auxenochlorella protothecoides and Chlorella spp. Chloroplast Genomes: Evidence for Endosymbiosis and Horizontal Virus-like Gene Transfer" Life 12, no. 3: 458. https://doi.org/10.3390/life12030458
APA StylePark, S. -H., Kyndt, J. A., & Brown, J. K. (2022). Comparison of Auxenochlorella protothecoides and Chlorella spp. Chloroplast Genomes: Evidence for Endosymbiosis and Horizontal Virus-like Gene Transfer. Life, 12(3), 458. https://doi.org/10.3390/life12030458