
The Impact of Genetic Polymorphisms in Glutamate-Cysteine Ligase, a Key Enzyme of Glutathione Biosynthesis, on Ischemic Stroke Risk and Brain Infarct Size

 ,
, 

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants and Clinical Examination
2.2. SNP Selection
2.3. Genetic Analysis
2.4. Biochemical Investigations
2.5. Statistical and Bioinformatics Analysis
3. Results
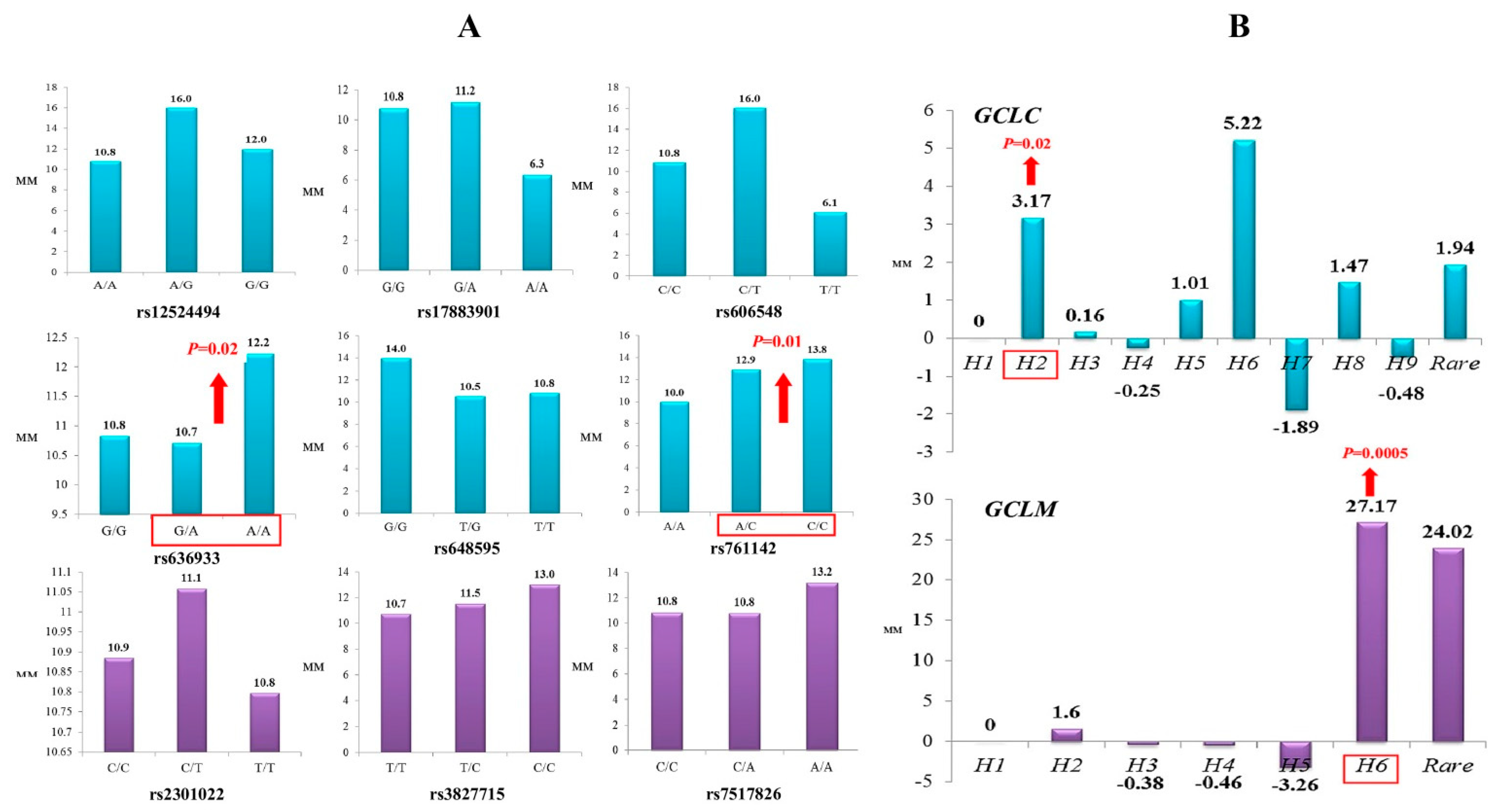
3.1. The Impact of the Studied Polymorphisms on the Risk of Ischemic Stroke and Brain Infarct Size
3.2. Replication Analysis for SNP-Disease Associations in Independent Populations
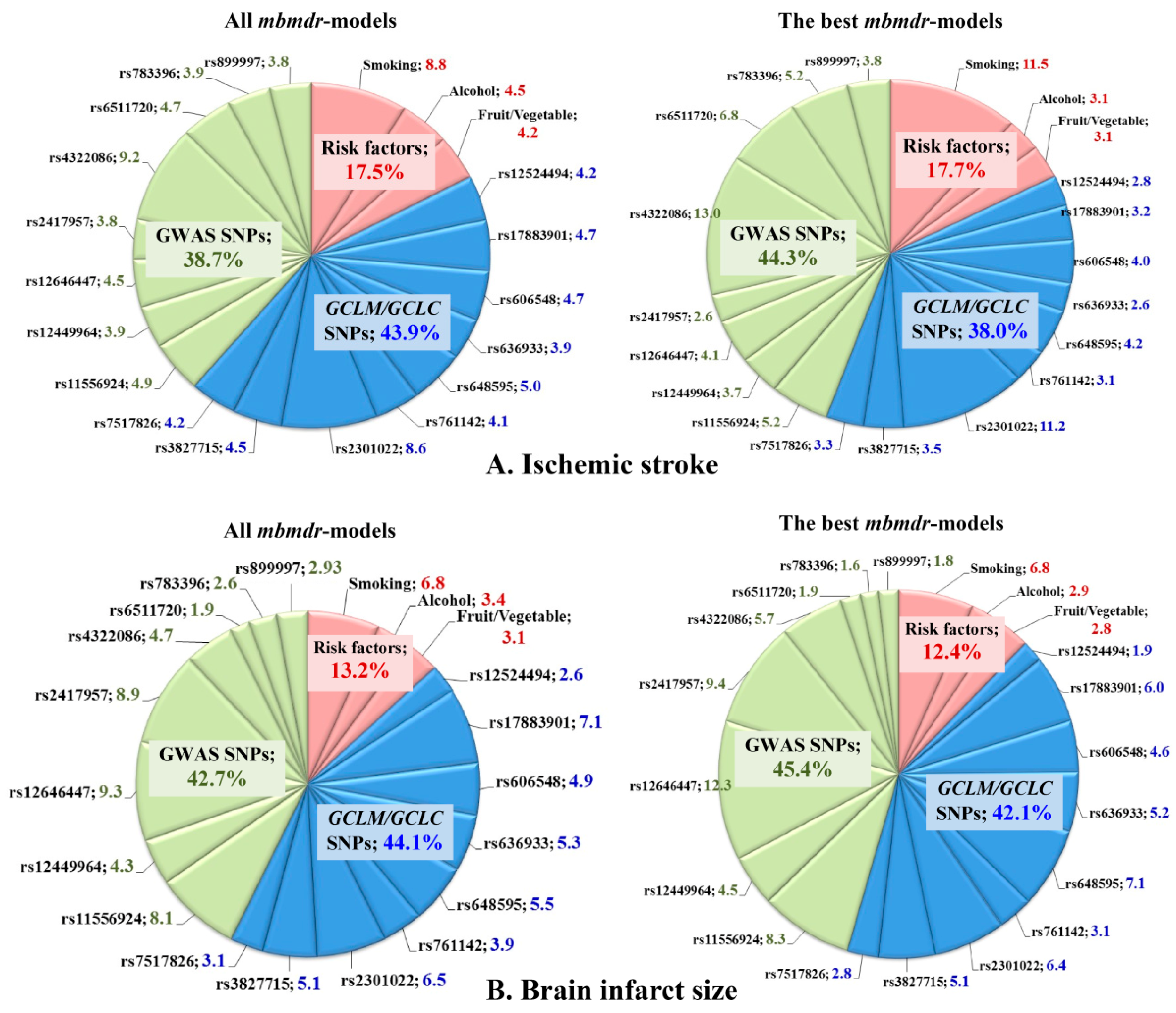
3.3. Gene–Gene and Gene–Environment Interactions, Ischemic Stroke Risk, and Brain InfarctSize
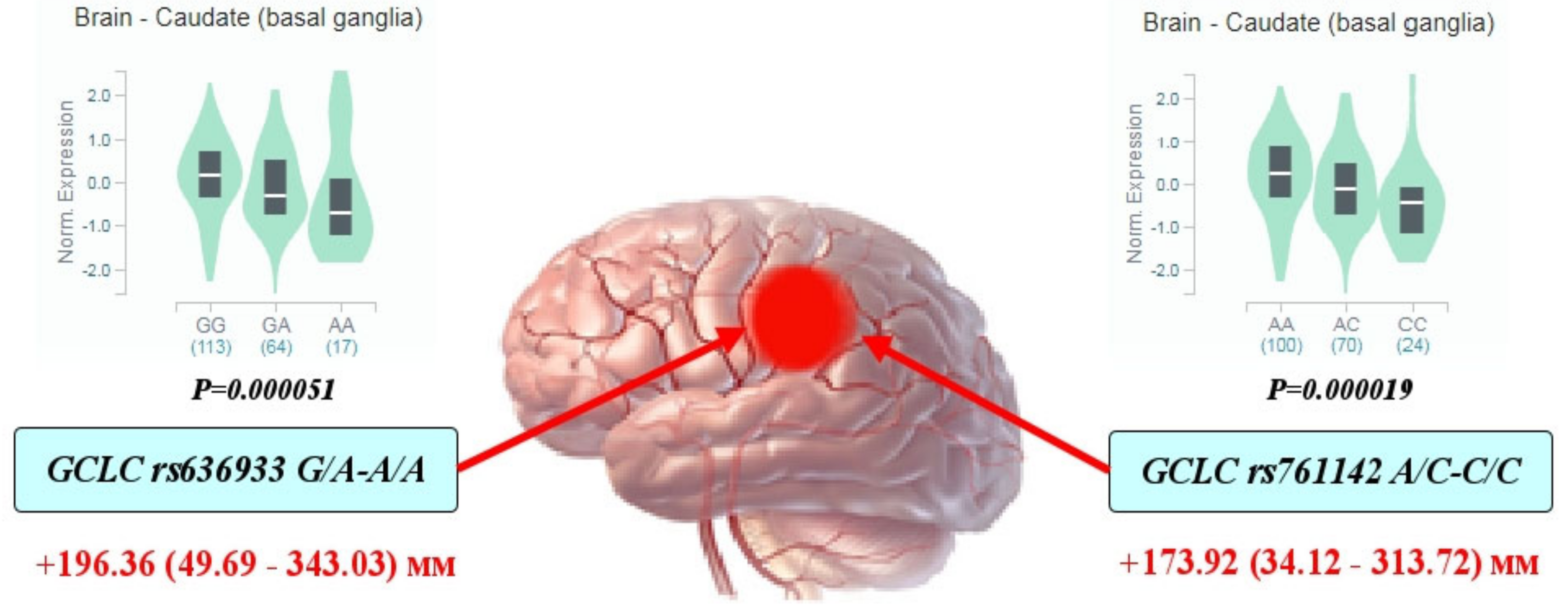
3.4. Functional Annotation of GCLC and GCLM Polymorphisms
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feigin, V.L. Stroke epidemiology in the developing world. Lancet 2005, 365, 2160–2161. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Colpo, G.D.; Venna, V.R.; McCullough, L.D.; Teixeira, A.L. Systematic Review on the Involvement of the Kynurenine Pathway in Stroke: Pre-clinical and Clinical Evidence. Front. Neurol. 2019, 10, 778. [Google Scholar] [CrossRef] [PubMed]
- Dichgans, M. Genetics of ischaemic stroke. Lancet Neurol. 2007, 6, 149–161. [Google Scholar] [CrossRef]
- Reis, J.; Giroud, M.; Kokubo, Y. Environmental Risk Factors for Stroke and Cardiovascular Disease. Encycl. Cardiovasc. Res. Med. 2017, 2018, 238–247. [Google Scholar] [CrossRef]
- Montaner, J.; Ramiro, L.; Simats, A.; Tiedt, S.; Makris, K.; Jickling, G.C.; Debette, S.; Sanchez, J.-C.; Bustamante, A. Multilevel omics for the discovery of biomarkers and therapeutic targets for stroke. Nat. Rev. Neurol. 2020, 16, 247–264. [Google Scholar] [CrossRef]
- Yang, H.; Jin, X.; Lam, C.W.K.; Yan, S.-K. Oxidative stress and diabetes mellitus. Clin. Chem. Lab. Med. (CCLM) 2011, 49, 1773–1782. [Google Scholar] [CrossRef]
- Vassalle, C.; Petrozzi, L.; Botto, N.; Andreassi, M.G.; Zucchelli, G.C. Oxidative stress and its association with coronary artery disease and different atherogenic risk factors. J. Intern. Med. 2004, 256, 308–315. [Google Scholar] [CrossRef]
- Allen, C.L.; Bayraktutan, U. Oxidative Stress and Its Role in the Pathogenesis of Ischaemic Stroke. Int. J. Stroke 2009, 4, 461–470. [Google Scholar] [CrossRef]
- Mury, P.; Chirico, E.N.; Mura, M.; Millon, A.; Canet-Soulas, E.; Pialoux, V. Oxidative Stress and Inflammation, Key Targets of Atherosclerotic Plaque Progression and Vulnerability: Potential Impact of Physical Activity. Sports Med. 2018, 48, 2725–2741. [Google Scholar] [CrossRef]
- Wu, G.; Fang, Y.-Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione Metabolism and Its Implications for Health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa-Díez, C.; Miguel, V.; Vallejo, S.; Sánchez, F.J.; Sandoval, E.; Blanco, E.; Cannata, P.; Peiro, C.; Sánchez-Ferrer, C.F.; Lamas, S. Role of glutathione biosynthesis in endothelial dysfunction and fibrosis. Redox Biol. 2018, 14, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, S.S.; Eshtehardi, P.; McDaniel, M.C.; Fike, L.V.; Jones, D.P.; Quyyumi, A.A.; Samady, H. The role of plasma aminothiols in the prediction of coronary microvascular dysfunction and plaque vulnerability. Atherosclerosis 2011, 219, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Bajic, V.P.; Van Neste, C.; Obradovic, M.; Zafirovic, S.; Radak, D.; Bajic, V.B.; Essack, M.; Isenovic, E.R. Glutathione “Redox Homeostasis” and Its Relation to Cardiovascular Disease. Oxidative Med. Cell. Longev. 2019, 2019, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.J.; Marchionni, L.; Andrews, C.E.; Norris, A.L.; Nucifora, L.G.; Wu, Y.C.; Wright, R.A.; Pevsner, J.; Ross, C.A.; Margolis, R.L.; et al. Analysis of differential gene expression mediated by clozapine in human postmortem brains. Schizophr. Res. 2016, 185, 58–66. [Google Scholar] [CrossRef]
- Kölsch, H.; Linnebank, M.; Lütjohann, D.; Jessen, F.; Wüllner, U.; Harbrecht, U.; Thelen, K.M.; Kreis, M.; Hentschel, F.; Schulz, A.; et al. Polymorphisms in glutathione S-transferase omega-1 and AD, vascular dementia, and stroke. Neurology 2004, 63, 2255–2260. [Google Scholar] [CrossRef]
- Türkanoğlu, A.; Demirdöğen, B.C.; Demirkaya, Ş.; Bek, S.; Adalı, O. Association analysis of GSTT1, GSTM1 genotype polymorphisms and serum total GST activity with ischemic stroke risk. Neurol. Sci. 2010, 31, 727–734. [Google Scholar] [CrossRef]
- Polonikov, A.; Vialykh, E.; Vasil’Eva, O.; Bulgakova, I.; Bushueva, O.; Illig, T.; Solodilova, M. Genetic Variation in Glutathione S-Transferase Genes and Risk of Nonfatal Cerebral Stroke in Patients Suffering from Essential Hypertension. J. Mol. Neurosci. 2012, 47, 511–513. [Google Scholar] [CrossRef]
- Wang, R.; Wang, Y.; Wang, J.; Yang, K. Association of glutathione S-transferase T1 and M1 gene polymorphisms with ischemic stroke risk in the Chinese Han population. Neural Regen. Res. 2012, 7, 1420–1427. [Google Scholar] [CrossRef]
- Bilgin, E.; Demirdöğen, B.C.; Özçelik, A.T.; Demirkaya, Ş.; Adalı, O. Association analysis of Glutathione S-transferase omega-1 and omega-2 genetic polymorphisms and ischemic stroke risk in a Turkish population. Neurol. Res. 2018, 41, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Voetsch, B.; Jin, R.C.; Bierl, C.; Benke, K.S.; Kenet, G.; Simioni, P.; Ottaviano, F.; Damasceno, B.P.; Annichino-Bizacchi, J.M.; Handy, D.E.; et al. Promoter polymorphisms in the plasma glutathione peroxidase (GPx-3) gene: A novel risk factor for arterial ischemic stroke among young adults and children. Stroke 2007, 38, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polonikov, A.V.; Vialykh, E.K.; Churnosov, M.; Illig, T.; Freidin, M.B.; Vasil'Eva, O.V.; Bushueva, O.; Ryzhaeva, V.N.; Bulgakova, I.V.; A Solodilova, M. The C718T polymorphism in the 3′-untranslated region of glutathione peroxidase-4 gene is a predictor of cerebral stroke in patients with essential hypertension. Hypertens. Res. 2011, 35, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Kiyohara, Y.; Kato, I.; Kitazono, T.; Tanizaki, Y.; Kubo, M.; Ueno, H.; Ibayashi, S.; Fujishima, M.; Iida, M. Relationship between plasma glutathione levels and cardiovascular disease in a defined population: The Hisayama study. Stroke 2004, 35, 2072–2077. [Google Scholar] [CrossRef] [Green Version]
- Cojocaru, I.M.; Cojocaru, M.; Sapira, V.; Ionescu, A. Evaluation of oxidative stress in patients with acute ischemic stroke. Rom. J. Intern. Med. 2013, 51, 97–106. [Google Scholar]
- Maksimova, M.Y.; Ivanov, A.V.; Virus, E.D.; Nikiforova, K.A.; Ochtova, F.R.; Suanova, E.T.; Kruglova, M.P.; Piradov, M.A.; Kubatiev, A.A. Impact of glutathione on acute ischemic stroke severity and outcome: Possible role of aminothiols redox status. Redox Rep. 2021, 26, 117–123. [Google Scholar] [CrossRef]
- Polonikov, A.; Rymarova, L.; Klyosova, E.; Volkova, A.; Azarova, I.; Bushueva, O.; Bykanova, M.; Bocharova, I.; Zhabin, S.; Churnosov, M.; et al. Matrix metalloproteinases as target genes for gene regulatory networks driving molecular and cellular pathways related to a multistep pathogenesis of cerebrovascular disease. J. Cell. Biochem. 2019, 120, 16467–16482. [Google Scholar] [CrossRef] [Green Version]
- Polonikov, A.V.; Bushueva, O.Y.; Bulgakova, I.V.; Freidin, M.B.; Churnosov, M.I.; Solodilova, M.A.; Shvetsov, Y.D.; Ivanov, V.P. A comprehensive contribution of genes for aryl hydrocarbon receptor signaling pathway to hypertension susceptibility. Pharm. Genom. 2017, 27, 57–69. [Google Scholar] [CrossRef] [Green Version]
- Klyosova, E.Y.; Azarova, I.E.; Sunyaykina, O.A.; Polonikov, A.V. Validity of a brief screener for environmental risk factors of age-related diseases using type 2 diabetes and coronary artery disease as examples. Res. Results Biomed. 2022, 8, 130–137. [Google Scholar] [CrossRef]
- Azarova, I.; Bushueva, O.; Konoplya, A.; Polonikov, A. Glutathione S-transferase genes and the risk of type 2 diabetes mellitus: Role of sexual dimorphism, gene-gene and gene-smoking interactions in disease susceptibility. J. Diabetes 2018, 10, 398–407. [Google Scholar] [CrossRef]
- Solé, X.; Guinó, E.; Valls, J.; Iniesta, R.; Moreno, V. SNPStats: A web tool for the analysis of association studies. Bioinformatics 2006, 22, 1928–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewontin, R.C. On measures of gametic disequilibrium. Genetics 1988, 120, 849–852. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D.; Hahn, L.W.; Roodi, N.; Bailey, L.R.; Dupont, W.D.; Parl, F.F.; Moore, J.H. Multifactor-Dimensionality Reduction Reveals High-Order Interactions among Estrogen-Metabolism Genes in Sporadic Breast Cancer. Am. J. Hum. Genet. 2001, 69, 138–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calle, M.L.; Urrea, V.; Vellalta, G.; Malats, N.; Steen, K.V. Improving strategies for detecting genetic patterns of disease susceptibility in association studies. Stat. Med. 2008, 27, 6532–6546. [Google Scholar] [CrossRef] [PubMed]
- Calle, M.L.; Urrea, V.; Malats, N.; Van Steen, K. mbmdr: An R package for exploring gene-gene interactions associated with binary or quantitative traits. Bioinformatics 2010, 26, 2198–2199. [Google Scholar] [CrossRef] [Green Version]
- Lazarenko, V.; Churilin, M.; Azarova, I.; Klyosova, E.; Bykanova, M.; Ob'Edkova, N.; Churnosov, M.; Bushueva, O.; Mal, G.; Povetkin, S.; et al. Comprehensive Statistical and Bioinformatics Analysis in the Deciphering of Putative Mechanisms by Which Lipid-Associated GWAS Loci Contribute to Coronary Artery Disease. Biomedicines 2022, 10, 259. [Google Scholar] [CrossRef]
- Xu, Z.; Taylor, J.A. SNPinfo: Integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res. 2009, 37, W600–W605. [Google Scholar] [CrossRef] [Green Version]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef] [Green Version]
- Aguet, F.; Barbeira, A.N.; Bonazzola, R.; Jo, B.; Kasela, S.; Liang, Y.; Parsana, P.; Aguet, F.; Battle, A.; Brown, A.; et al. The GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.-J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef]
- Zheng, Z.; Huang, D.; Wang, J.; Zhao, K.; Zhou, Y.; Guo, Z.; Zhai, S.; Xu, H.; Cui, H.; Yao, H.; et al. QTLbase: An integrative resource for quantitative trait loci across multiple human molecular phenotypes. Nucleic Acids Res. 2019, 48, D983–D991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, A.D.; Oscanoa, J.; Wang, J.; Nagano, A.; Lemoine, N.R.; Chelala, C. SNPnexus: Assessing the functional relevance of genetic variation to facilitate the promise of precision medicine. Nucleic Acids Res. 2018, 46, W109–W113. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. TRANSFAC and its module TRANSCompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.; Hudson, R.; Harrison, C.; Craven, M.; Keleş, S. atSNP Search: A web resource for statistically evaluating influence of human genetic variation on transcription factor binding. Bioinformatics 2018, 35, 2657–2659. [Google Scholar] [CrossRef] [PubMed]
- Dugourd, A.; Saez-Rodriguez, J. Footprint-based functional analysis of multiomic data. Curr. Opin. Syst. Biol. 2019, 15, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Beck, S. Making multi-omics data accessible to researchers. Sci. Data 2019, 6, 251. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Man, B.; Baum, L.; Fu, Y.; Chan, Y.; Lam, W.; Hui, C.; Leung, W.; Wong, K. Genetic polymorphisms of Chinese patients with ischemic stroke and concurrent stenoses of extracranial and intracranial vessels. J. Clin. Neurosci. 2010, 17, 1244–1247. [Google Scholar] [CrossRef]
- Campolo, J.; Penco, S.; Bianchi, E.; Colombo, L.; Parolini, M.; Caruso, R.; Sedda, V.; Patrosso, M.C.; Cighetti, G.; Marocchi, A.; et al. Glutamate-cysteine ligase polymorphism, hypertension, and male sex are associated with cardiovascular events. Biochemical and genetic characterization of Italian subpopulation. Am. Hear. J. 2007, 154, 1123–1129. [Google Scholar] [CrossRef]
- Baum, L.; Chen, X.; Cheung, W.S.; Cheung, C.K.A.; Cheung, L.W.; Chiu, K.F.P.; Wen, H.M.; Poon, P.; Woo, K.S.; Ng, H.K.; et al. Polymorphisms and Vascular Cognitive Impairment After Ischemic Stroke. J. Geriatr. Psychiatry Neurol. 2007, 20, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.-I.; Kugiyama, K.; Sugiyama, S.; Miyamoto, S.; Koide, S.-I.; Fukushima, H.; Honda, O.; Yoshimura, M.; Ogawa, H. Polymorphism in the 5′-Flanking Region of Human Glutamate-Cysteine Ligase Modifier Subunit Gene Is Associated With Myocardial Infarction. Circulation 2002, 105, 2968–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koide, S.-I.; Kugiyama, K.; Sugiyama, S.; Nakamura, S.-I.; Fukushima, H.; Honda, O.; Yoshimura, M.; Ogawa, H. Association of polymorphism in glutamate-cysteine ligase catalytic subunit gene with coronary vasomotor dysfunction and myocardial infarction. J. Am. Coll. Cardiol. 2003, 41, 539–545. [Google Scholar] [CrossRef] [Green Version]
- Traylor, M.; Malik, R.; Nalls, M.A.; Cotlarciuc, I.; Radmanesh, F.; Thorleifsson, G.; Hanscombe, K.B.; Langefeld, C.; Saleheen, D.; Rost, N.S.; et al. Genetic variation at 16q24.2 is associated with small vessel stroke. Ann. Neurol. 2016, 81, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dichgans, M.; Malik, R.; König, I.; Rosand, J.; Clarke, R.; Gretarsdottir, S.; Thorleifsson, G.; Mitchell, B.; Assimes, T.L.; Levi, C.; et al. Shared genetic susceptibility to ischemic stroke and coronary artery disease: A genome-wide analysis of common variants. Stroke 2014, 45, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Pirruccello, J.P.; Bick, A.; Wang, M.; Chaffin, M.; Friedman, S.; Yao, J.; Guo, X.; Venkatesh, B.A.; Taylor, K.D.; Post, W.S.; et al. Analysis of cardiac magnetic resonance imaging in 36,000 individuals yields genetic insights into dilated cardiomyopathy. Nat. Commun. 2020, 11, 2254. [Google Scholar] [CrossRef]
- Kotlęga, D.; Gołąb-Janowska, M.; Masztalewicz, M.; Ciećwież, S.; Nowacki, P. The emotional stress and risk of ischemic stroke. Neurol. I Neurochir. Pol. 2016, 50, 265–270. [Google Scholar] [CrossRef]
- Mostofsky, E.; Laier, E.; Levitan, E.B.; Rosamond, W.D.; Schlaug, G.; Mittleman, M.A. Physical Activity and Onset of Acute Ischemic Stroke: The Stroke Onset Study. Am. J. Epidemiol. 2010, 173, 330–336. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Ye, W.; Adami, H.-O.; Weiderpass, E. Stroke Incidence in Women under 60 Years of Age Related to Alcohol Intake and Smoking Habit. Cerebrovasc. Dis. 2008, 25, 517–525. [Google Scholar] [CrossRef]
- Mukamal, K.J.; Ascherio, A.; Mittleman, M.; Conigrave, K.M.; Camargo, C.A.; Kawachi, I.; Stampfer, M.J.; Willett, W.C.; Rimm, E.B. Alcohol and Risk for Ischemic Stroke in Men: The Role of Drinking Patterns and Usual Beverage. Ann. Intern. Med. 2005, 142, 11–19. [Google Scholar] [CrossRef]
- Joshipura, K.; Ascherio, A.; Manson, J.E.; Stampfer, M.J.; Rimm, E.B.; Speizer, F.E.; Hennekens, C.H.; Spiegelman, D.; Willett, W.C. Fruit and Vegetable Intake in Relation to Risk of Ischemic Stroke. JAMA 1999, 282, 1233–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persky, H. Glutathione Metabolism in Men under Psychological Stress. Psychosom. Med. 1954, 16, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.C.; Raiford, D.S.; Mallat, A. Effects of Ethanol on Glutathione Metabolism. In Liver Pathology and Alcohol; Watson, R.R., Ed.; Humana Press: Totowa, NJ, USA, 1991; Volume 2. [Google Scholar]
- Vogt, B.L.; Richie, J.P. Glutathione depletion and recovery after acute ethanol administration in the aging mouse. Biochem. Pharmacol. 2007, 73, 1613–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazzini, C.; Rossetti, V.; Civello, D.A.; Sassone, F.; Vezzoli, V.; Persani, L.; Tiberio, L.; Lanata, L.; Bagnasco, M.; Paulmichl, M.; et al. Short- and Long- Term Effects of Cigarette Smoke Exposure on Glutathione Homeostasis in Human Bronchial Epithelial Cells. Cell. Physiol. Biochem. 2013, 32, 129–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Toorn, M.; Vries, M.P.S.-D.; Slebos, D.-J.; De Bruin, H.G.; Abello, N.; Van Oosterhout, A.J.M.; Bischoff, R.; Kauffman, H.F. Cigarette smoke irreversibly modifies glutathione in airway epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2007, 293, L1156–L1162. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Garavaglia, M.L.; Colombo, G.; Astori, E.; Lionetti, M.C.; La Porta, C.A.; Santucci, A.; Rossi, R.; Giustarini, D.; Milzani, A. Cigarette smoke and glutathione: Focus on in vitro cell models. Toxicol. Vitr. 2020, 65, 104818. [Google Scholar] [CrossRef]
- Sen, C.K. Glutathione: A key role in skeletal muscle metabolism. In Oxidative Stress in Skeletal Muscle; Reznick, A.Z., Packer, L., Sen, C.K., Holloszy, J.O., Jackson, M.J., Eds.; Birkhäuser: Basel, Switzerland, 1998. [Google Scholar]
- Nyberg, M.; Mortensen, S.P.; Cabo, H.; Gomez-Cabrera, M.-C.; Viña, J.; Hellsten, Y. Roles of sedentary aging and lifelong physical activity in exchange of glutathione across exercising human skeletal muscle. Free Radic. Biol. Med. 2014, 73, 166–173. [Google Scholar] [CrossRef]
- Minich, D.M.; Brown, B.I. A Review of Dietary (Phyto)Nutrients for Glutathione Support. Nutrients 2019, 11, 2073. [Google Scholar] [CrossRef] [Green Version]
- Kolesnichenko, L.S.; Mantorova, N.S.; A Shapiro, L.; A Ol'Khovskiĭ, I.; Baron, A.V. Effect of emotional stress on the activity of enzymes of glutathione metabolism. Vopr. Med. Khim. 1987, 33, 85–88. [Google Scholar]
- Herbet, M.; Korga, A.; Gawrońska-Grzywacz, M.; Izdebska, M.; Piątkowska-Chmiel, I.; Poleszak, E.; Wróbel, A.; Matysiak, W.; Jodłowska-Jędrych, B.; Dudka, J. Chronic Variable Stress Is Responsible for Lipid and DNA Oxidative Disorders and Activation of Oxidative Stress Response Genes in the Brain of Rats. Oxidative Med. Cell. Longev. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Corcoba, A.; Gruetter, R.; Do, K.Q.; Duarte, J.M. Social isolation stress and chronic glutathione deficiency have a common effect on the glutamine-to-glutamate ratio andmyo-inositol concentration in the mouse frontal cortex. J. Neurochem. 2017, 142, 767–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragsted, L.O.; Pedersen, A.; Hermetter, A.; Basu, S.; Hansen, M.; Haren, G.R.; Kall, M.; Breinholt, V.; Castenmiller, J.J.; Stagsted, J.; et al. The 6-a-day study: Effects of fruit and vegetables on markers of oxidative stress and antioxidative defense in healthy nonsmokers. Am. J. Clin. Nutr. 2004, 79, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, C.C.; Backos, D.S.; Mohar, I.; White, C.C.; Forman, H.J.; Kavanagh, T.J. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Asp. Med. 2009, 30, 86–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, A.C.; Mulcahy, R.T. Regulation of gamma-glutamylcysteine synthetase subunit gene expression: Insights into transcriptional control of antioxidant defenses. Free Radic. Res. 2000, 32, 281–301. [Google Scholar] [CrossRef]
- Aoyama, K.; Watabe, M.; Nakaki, T. Regulation of Neuronal Glutathione Synthesis. J. Pharmacol. Sci. 2008, 108, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Bocharova, I.A. An association study of three polymorphisms in the glutathione synthase (GSS) gene with the risk of ischemic stroke. Res. Results Biomed. 2020, 6, 476–487. [Google Scholar] [CrossRef]
- Bocharova, J.A.; Azarova, J.E.; Klyosova, E.Y.; Drozdova, E.L.; Solodilova, M.A.; Polonikov, A.V. Gene of gamma-glutamylcyclotransferase, a key enzyme of glutathione catabolism, and predisposition to ischemic stroke: Association analysis and functional annotation of gene polymorphisms. Med. Genet. 2020, 19, 32–39. [Google Scholar] [CrossRef]
- Bocharova, Y. Associations between glutamate cysteine ligase catalytic subunit gene polymorphisms and clinical characteristics of ischemic stroke. Bull. Russ. State Med. Univ. 2021, 1, 19–23. [Google Scholar] [CrossRef]
- Callegari, A.; Liu, Y.; White, C.C.; Chait, A.; Gough, P.; Raines, E.W.; Cox, D.; Kavanagh, T.J.; Rosenfeld, M.E. Gain and loss of function for glutathione synthesis: Impact on advanced atherosclerosis in apolipoprotein E-deficient mice. Arter. Thromb. Vasc. Biol. 2011, 31, 2473–2482. [Google Scholar] [CrossRef] [Green Version]
- Busu, C.; Atanasiu, V.; Caldito, G.; Aw, T.Y. Influence of GSH synthesis inhibition on temporal distribution of NAD+/NADH during vascular endothelial cells proliferation. J. Med. Life 2014, 7, 611–618. [Google Scholar]
- Lapenna, D.; De Gioia, S.; Ciofani, G.; Mezzetti, A.; Ucchino, S.; Calafiore, A.M.; Napolitano, A.M.; Di Ilio, C.; Cuccurullo, F. Glutathione-Related Antioxidant Defenses in Human Atherosclerotic Plaques. Circulation 1998, 97, 1930–1934. [Google Scholar] [CrossRef] [PubMed]
- Malekmohammad, K.; Sewell, R.D.E.; Rafieian-Kopaei, M. Antioxidants and Atherosclerosis: Mechanistic Aspects. Biomolecules 2019, 9, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K.; Newby, D.E.; Rahman, I.; Megson, I.L. Depressed glutathione synthesis precedes oxidative stress and atherogenesis in Apo-E−/− mice. Biochem. Biophys. Res. Commun. 2005, 338, 1368–1373. [Google Scholar] [CrossRef]
- Sleptsov, A.A.; Nazarenko, M.S.; Lebedev, I.N.; Skriabin, N.A.; Frolov, A.V.; Popov, V.A.; Barbarash, L.S.; Puzyrev, V.P. Somatic genome variations in vascular tissues and peripheral blood leukocytes in patients with atherosclerosis. Genetika 2014, 50, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Bushueva, O.; Barysheva, E.; Markov, A.; Belykh, A.; Koroleva, I.; Churkin, E.; Polonikov, A.; Ivanov, V.; Nazarenko, M. DNA Hypomethylation of the MPO Gene in Peripheral Blood Leukocytes Is Associated with Cerebral Stroke in the Acute Phase. J. Mol. Neurosci. 2021, 71, 1914–1932. [Google Scholar] [CrossRef]
- Bots, M.L.; Hofman, A.; De Jong, P.T.; Grobbee, D.E. Common carotid intima-media thickness as an indicator of atherosclerosis at other sites of the carotid artery the Rotterdam Study. Ann. Epidemiol. 1996, 6, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Iwakiri, T.; Yano, Y.; Sato, Y.; Hatakeyama, K.; Marutsuka, K.; Fujimoto, S.; Kitamura, K.; Kario, K.; Asada, Y. Usefulness of carotid intima-media thickness measurement as an indicator of generalized atherosclerosis: Findings from autopsy analysis. Atherosclerosis 2012, 225, 359–362. [Google Scholar] [CrossRef]
- Schutte, R.; Schutte, A.E.; Huisman, H.; Van Rooyen, J.M.; Malan, L.; Péter, S.; Fourie, C.M.T.; Van Der Westhuizen, F.H.; Louw, R.; Botha, C.A.; et al. Blood glutathione and subclinical atherosclerosis in African men: The SABPA Study. Am. J. Hypertens. 2009, 22, 1154–1159. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.; Hazama, M.; Urata, Y.; Goto, S.; Horiuchi, S.; Sumikawa, K.; Kondo, T. Protective role of glutathione synthesis in response to oxidized low density lipoprotein in human vascular endothelial cells. Free Radic. Biol. Med. 1999, 26, 589–602. [Google Scholar] [CrossRef]
- Prasad, A.; Andrews, N.P.; A Padder, F.; Husain, M.; A Quyyumi, A. Glutathione reverses endothelial dysfunction and improves nitric oxide bioavailability. J. Am. Coll. Cardiol. 1999, 34, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Salemi, G.; Gueli, M.C.; D'Amelio, M.; Saia, V.; Mangiapane, P.; Aridon, P.; Ragonese, P.; Lupo, I. Blood levels of homocysteine, cysteine, glutathione, folic acid, and vitamin B12 in the acute phase of atherothrombotic stroke. Neurol. Sci. 2009, 30, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Mizui, T.; Kinouchi, H.; Chan, P.H. Depletion of brain glutathione by buthionine sulfoximine enhances cerebral ischemic injury in rats. Am. J. Physiol. Circ. Physiol. 1992, 262, H313–H317. [Google Scholar] [CrossRef] [PubMed]
- Juurlink, B.; Schültke, E.; Hertz, L. Glutathione release and catabolism during energy substrate restriction in astrocytes. Brain Res. 1996, 710, 229–233. [Google Scholar] [CrossRef]
- Song, J.; Park, J.; Oh, Y.; Lee, J.E. Glutathione Suppresses Cerebral Infarct Volume and Cell Death after Ischemic Injury: Involvement of FOXO3 Inactivation and Bcl2 Expression. Oxid. Med. Cell. Longev. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Wang, B.; Aw, T.Y.; Stokes, K.Y. The protection conferred against ischemia-reperfusion injury in the diabetic brain by N-acetylcysteine is associated with decreased dicarbonyl stress. Free Radic. Biol. Med. 2016, 96, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Kahl, A.; Stepanova, A.; Konrad, C.; Anderson, C.; Manfredi, G.; Zhou, P.; Iadecola, C.; Galkin, A. Critical Role of Flavin and Glutathione in Complex I–Mediated Bioenergetic Failure in Brain Ischemia/Reperfusion Injury. Stroke 2018, 49, 1223–1231. [Google Scholar] [CrossRef]
- Yoshida, M.; Mikami, T.; Higashi, K.; Saiki, R.; Mizoi, M.; Fukuda, K.; Nakamura, T.; Ishii, I.; Nishimura, K.; Toida, T.; et al. Inverse correlation between stroke and urinary 3-hydroxypropyl mercapturic acid, an acrolein-glutathione metabolite. Clin. Chim. Acta 2012, 413, 753–759. [Google Scholar] [CrossRef]
- Paterson, P.G.; Juurlink, B.H.J. Nutritional regulation of glutathione in stroke. Neurotox. Res. 1999, 1, 99–112. [Google Scholar] [CrossRef]
- Bansal, M.; Kaushal, N. Cell signalling and gene regulation by oxidative stress. In Oxidative Stress Mechanisms and Their Modulation; Bansal, M., Kaushal, N., Eds.; Springer: New Delhi, India, 2014. [Google Scholar]
- Sedlak, T.W.; Paul, B.D.; Parker, G.M.; Hester, L.D.; Snowman, A.M.; Taniguchi, Y.; Kamiya, A.; Snyder, S.H.; Sawa, A. The glutathione cycle shapes synaptic glutamate activity. Proc. Natl. Acad. Sci. USA 2019, 116, 2701–2706. [Google Scholar] [CrossRef] [Green Version]
- Kolesnichenko, L.S.; Kulinskiĭ, V.I.; Shprakh, V.V.; Bardymov, V.V.; Verlan, N.V.; Gubina, L.P.; Pensionerova, G.A.; Sergeeva, M.P.; Stanevich, L.M.; Filippova, G.T. The blood glutathione system in cerebral vascular diseases and its treatment with alpha-lipoic acid. Zhurnal Nevrol. I Psikhiatrii Im. SS Korsakova 2008, 108, 36–40. [Google Scholar]
- Anderson, M.F.; Sims, N.R. The effects of focal ischemia and reperfusion on the glutathione content of mitochondria from rat brain subregions. J. Neurochem. 2002, 81, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Maher, P. The effects of stress and aging on glutathione metabolism. Ageing Res. Rev. 2005, 4, 288–314. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Watanabe, K.; Ishibashi, M.; Saiki, R.; Kuni, K.; Nishimura, K.; Toida, T.; Kashiwagi, K.; Igarashi, K. Aggravation of brain infarction through an increase in acrolein production and a decrease in glutathione with aging. Biochem. Biophys. Res. Commun. 2016, 473, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Pizzurro, D.M.; Dao, K.; Costa, L.G. Astrocytes protect against diazinon- and diazoxon-induced inhibition of neurite outgrowth by regulating neuronal glutathione. Toxicology 2014, 318, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, C.; Winnefeld, K.; Streck, S.; Roskos, M.; Haberl, R.L. Antioxidant Status in Acute Stroke Patients and Patients at Stroke Risk. Eur. Neurol. 2004, 51, 157–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdo, J.; Schubert, D.; Maher, P. Glutathione production is regulated via distinct pathways in stressed and non-stressed cortical neurons. Brain Res. 2008, 1189, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Roth, T.; Nayak, D.; Atanasijevic, T.; Koretsky, A.; Latour, L.L.; McGAVERN, D.B. Transcranial amelioration of inflammation and cell death after brain injury. Nature 2013, 505, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef] [Green Version]
- Howarth, C.; Sutherland, B.A.; Choi, H.B.; Martin, C.; Lind, B.L.; Khennouf, L.; LeDue, J.M.; Pakan, J.M.; Ko, R.W.; Ellis-Davies, G.; et al. A Critical Role for Astrocytes in Hypercapnic Vasodilation in Brain. J. Neurosci. 2017, 37, 2403–2414. [Google Scholar] [CrossRef]
- Khan, M.; Dhammu, T.; Matsuda, F.; Baarine, M.; Dhindsa, T.; Singh, I.; Singh, A. Promoting endothelial function by S-nitrosoglutathione through the HIF-1α/VEGF pathway stimulates neurorepair and functional recovery following experimental stroke in rats. Drug Des. Dev. Ther. 2015, 9, 2233–2247. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, H.F. Molecular and cellular aspects of thiol-disulfide exchange. Adv. Enzymol. Relat. Areas Mol. Biol. 1990, 63, 69–172. [Google Scholar] [PubMed]
- Okumura, M.; Saiki, M.; Yamaguchi, H.; Hidaka, Y. Acceleration of disulfide-coupled protein folding using glutathione derivatives. FEBS J. 2011, 278, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Sinskey, A.; Lodish, H. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Paschen, W.; Doutheil, J. Disturbances of the Functioning of Endoplasmic Reticulum: A Key Mechanism Underlying Neuronal Cell Injury? J. Cereb. Blood Flow Metab. 1999, 19, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Yuan, M.; Guo, Y.-S.; Shen, X.-Y.; Gao, Z.-K.; Bi, X. Mechanism of Endoplasmic Reticulum Stress in Cerebral Ischemia. Front. Cell Neurosci. 2021, 15, 704334. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; Mazzon, E.; Costantino, G.; Serraino, I.; Dugo, L.; Calabrò, G.; Cucinotta, G.; De Sarro, A.; Caputi, A.P. Beneficial effects ofn-acetylcysteine on ischaemic brain injury. J. Cereb. Blood Flow Metab. 2000, 130, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Uemura, T.; Watanabe, K.; Ko, K.; Higashi, K.; Kogure, N.; Kitajima, M.; Takayama, H.; Takao, K.; Sugita, Y.; Sakamoto, A.; et al. Protective Effects of Brain Infarction by N-Acetylcysteine Derivatives. Stroke 2018, 49, 1727–1733. [Google Scholar] [CrossRef]
- Sabetghadam, M.; Mazdeh, M.; Abolfathi, P.; Mohammadi, Y.; Mehrpooya, M. Evidence for a Beneficial Effect of Oral N-acetylcysteine on Functional Outcomes and Inflammatory Biomarkers in Patients with Acute Ischemic Stroke. Neuropsychiatr. Dis. Treat. 2020, 16, 1265–1278. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Baseline and Clinical Characteristics | Controls (n = 688) | IS Patients (n = 600) | p-Value | ||
|---|---|---|---|---|---|
| Age, M ± S.D. | 60.8 ± 7.5 | 61.1 ± 9.8 | 0.59 | ||
| Sex, n (%) | Males | 366 (53.2) | 330 (55.0) | 0.52 | |
| Females | 322 (46.8) | 270 (45.0) | |||
| BMI (kg/m2), M ± S.D. | 24.6 ± 3.8 | 25.2 ± 4.2 | 0.11 | ||
| Brain infarct size (mm in maximal diameter), Me (Q1–Q3) | - | 10.8 (5.0–23.9) | - | ||
| Hypertension | - | 586 (97.7) | - | ||
| Coronary artery disease | - | 49 (8.2) | - | ||
| Diabetes mellitus | - | 52 (8.7) | - | ||
| Smoking status * | Ever | 221 (32.8) | 265 (44.2) | <0.0001 | |
| Never | 452 (67.2) | 335 (55.8) | |||
| Alcohol intake * | Abuse | 25 (10.0) | 116 (19.3) | 0.001 | |
| Low/moderate | 226 (90.0) | 484 (80.7) | |||
| Fruits/vegetables intake * | Low | 100 (39.2) | 283 (47.3) | 0.29 | |
| High/moderate | 155 (60.8) | 315 (52.7) | |||
| Oxidized glutathione (GSSG) in plasma (μmol/L), Me (Q1–Q3) * | 1.93 (0.84–5.75) | 1.31 (0.46–3.52) | 0.008 | ||
| Reactive oxygen species (ROS) in plasma (μmol/L), Me (Q1–Q3) * | 2.47 (1.98–3.69) | 3.41 (2.43–4.21) | 0.004 | ||
| Gene (SNP ID) | Genotype, Allele | n (%) | p-Value | corOR (95% CI) * | |
|---|---|---|---|---|---|
| Controls (n = 688) | IS Patients (n = 600) | ||||
| GCLC A > G (rs12524494) | A/A | 543 (93.6) | 550 (93.4) | 0.98 | 1.00 |
| A/G | 33 (5.7) | 35 (5.9) | 1.05 (0.64–1.72) | ||
| G/G | 4 (0.7) | 4 (0.7) | 1.02 (0.25–4.10) | ||
| G | 0.035 | 0.037 | 0.88 | 1.03 (0.67–1.59) | |
| GCLC G > A (rs17883901) | G/G | 585 (86.2) | 493 (84.4) | 0.30 | 1.00 |
| G/A | 90 (13.2) | 83 (14.2) | 1.10 (0.80–1.52) | ||
| A/A | 4 (0.6) | 8 (1.4) | 2.38 (0.71–7.95) | ||
| A | 0.072 | 0.085 | 0.24 | 1.19 (0.89–1.59) | |
| GCLC C > T (rs606548) | C/C | 625 (94) | 496 (93.2) | 0.88 | 1.00 |
| C/T | 38 (5.7) | 34 (6.4) | 1.12 (0.69–1.81) | ||
| T/T | 2 (0.3) | 2 (0.4) | 1.26 (0.18–8.97) | ||
| T | 0.032 | 0.036 | 0.58 | 1.14 (0.73–1.77) | |
| GCLC G > A (rs636933) | G/G | 421 (62.7) | 370 (64.5) | 0.79 | 1.00 |
| G/A | 216 (32.2) | 178 (31) | 0.94 (0.74–1.20) | ||
| A/A | 34 (5.1) | 26 (4.5) | 0.87 (0.51–1.48) | ||
| A | 0.212 | 0.200 | 0.49 | 0.93 (0.77–1.13) | |
| GCLC G > T (rs648595) | G/G | 124 (18.2) | 82 (14.1) | 0.049 | 1.00 |
| G/T | 335 (49.2) | 323 (55.4) | 1.20 (0.93–1.54) | ||
| T/T | 222 (32.6) | 178 (30.5) | 0.91 (0.72–1.15) | ||
| T | 0.572 | 0.582 | 0.60 | 1.04 (0.89–1.22) | |
| GCLC A > C (rs761142) | A/A | 388 (57.2) | 331 (57.3) | 0.92 | 1.00 |
| A/C | 255 (37.6) | 220 (38.1) | 1.01 (0.80–1.27) | ||
| C/C | 35 (5.2) | 27 (4.7) | 0.90 (0.54–1.53) | ||
| C | 0.240 | 0.237 | 0.88 | 0.99 (0.82–1.18) | |
| GCLM C > T (rs2301022) | C/C | 344 (51) | 286 (51) | <0.0001 | 1.00 |
| C/T | 251 (37.2) | 249 (44.4) | 1.19 (0.94–1.51) | ||
| T/T | 80 (11.8) | 26 (4.6) | 0.39 (0.24–0.62) | ||
| T | 0.304 | 0.268 | 0.048 | 0.84 (0.70–1.00) | |
| GCLM T > C (rs3827715) | T/T | 348 (52.5) | 293 (52.2) | 0.31 | 1.00 |
| T/C | 258 (38.9) | 232 (41.4) | 1.07 (0.85–1.36) | ||
| C/C | 57 (8.6) | 36 (6.4) | 0.76 (0.48–1.18) | ||
| C | 0.281 | 0.271 | 0.60 | 0.95 (0.80–1.14) | |
| GCLM C > A (rs7517826) | C/C | 254 (38.7) | 207 (37.2) | 0.62 | 1.00 |
| C/A | 306 (46.6) | 275 (49.4) | 1.10 (0.86–1.41) | ||
| A/A | 96 (14.6) | 75 (13.5) | 0.96 (0.67–1.37) | ||
| A | 0.380 | 0.382 | 0.92 | 1.01 (0.86–1.19) | |
| Haplotypes | SNPs | Frequency | p-Value | adjOR (95%CI) 2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| rs12524494 | rs636933 | rs648595 | rs761142 | rs606548 | rs17883901 | Healthy Controls | IS Patients | |||
| GCLC Haplotype Frequencies (n = 1288) | ||||||||||
| H1 | A | G | T | A | C | G | 0.5383 | 0.5388 | - | 1.00 |
| H2 | A | A | G | C | C | G | 0.1536 | 0.1583 | 0.87 | 1.02 (0.80–1.29) |
| H3 | A | G | G | A | C | G | 0.1575 | 0.1432 | 0.45 | 0.91 (0.72–1.16) |
| H4 | G | G | G | C | T | G | 0.0325 | 0.0369 | 0.63 | 1.11 (0.72–1.72) |
| H5 | A | G | T | A | C | A | 0.0278 | 0.0394 | 0.17 | 1.42 (0.86–2.35) |
| H6 | A | A | G | C | C | A | 0.0333 | 0.0222 | 0.25 | 0.72 (0.42–1.25) |
| H7 | A | A | G | A | C | G | 0.0253 | 0.0162 | 0.28 | 0.73 (0.42–1.29) |
| H8 | A | G | G | C | C | G | 0.0081 | 0.0152 | 0.024 | 3.37 (1.18–9.62) |
| H9 | A | G | G | A | C | A | 0.0060 | 0.0180 | 0.19 | 1.75 (0.76–4.03) |
| Rare 1 | * | * | * | * | * | * | 0.0176 | 0.0118 | 0.16 | 0.56 (0.25–1.27) |
| Global haplotype association p-value: 0.11 | ||||||||||
| GCLM haplotype frequencies estimation (n = 1285) | ||||||||||
| Haplotypes | rs7517826 | rs3827715 | rs2301022 | Healthy Controls | IS Patients | p-Value | adjOR (95%CI) 2 | |||
| H1 | C | T | C | 0.3536 | 0.3982 | - | 1.00 | |||
| H2 | A | C | C | 0.2660 | 0.2497 | 0.097 | 0.83 (0.67–1.03) | |||
| H3 | C | T | T | 0.2692 | 0.2294 | 0.016 | 0.76 (0.61–0.95) | |||
| H4 | A | T | C | 0.0757 | 0.0835 | 0.86 | 0.97 (0.68–1.37) | |||
| H5 | A | C | T | 0.0194 | 0.0249 | 0.72 | 1.14 (0.55–2.36) | |||
| H6 | A | T | T | 0.0153 | 0.0123 | 0.48 | 0.71 (0.28–1.84) | |||
| Rare 1 | * | * | * | 0.0008 | 0.0020 | 0.52 | 2.29 (0.18–28.8) | |||
| Global haplotype association p-value: 0.27 | ||||||||||
| GCLM Haplotype Frequencies Estimation in NonSmokers (n = 785) | |||||||
|---|---|---|---|---|---|---|---|
| Haplotypes | rs7517826 | rs3827715 | rs2301022 | Healthy Controls | IS Patients | p-Value | adjOR (95%CI) 2 |
| H1 | C | T | C | 0.3266 | 0.4104 | - | 1.00 |
| H2 | A | C | C | 0.2852 | 0.2447 | 0.0047 | 0.65 (0.49–0.88) |
| H3 | C | T | T | 0.2845 | 0.2095 | 3 × 10−4 | 0.58 (0.43–0.78) |
| H4 | A | T | C | 0.0782 | 0.0897 | 0.59 | 0.89 (0.57–1.37) |
| H5 | A | C | T | 0.0212 | 0.0327 | 0.56 | 1.29 (0.56–2.96) |
| H6 | A | T | T | 0.0029 | 0.0095 | 0.30 | 2.77 (0.40–19.16) |
| Rare 1 | * | * | * | 0.0014 | 0.0035 | ||
| Global haplotype association p-value: 0.0027 | |||||||
| GCLM haplotype frequencies estimation in smokers (n = 485) | |||||||
| H1 | C | T | C | 0.4079 | 0.3842 | - | 1.00 |
| H2 | A | C | C | 0.2307 | 0.2542 | 0.52 | 1.13 (0.78–1.64) |
| H3 | C | T | T | 0.2384 | 0.2531 | 0.46 | 1.15 (0.80–1.65) |
| H4 | A | T | C | 0.0678 | 0.0767 | 0.59 | 1.19 (0.64–2.23) |
| H5 | A | C | T | 0.0194 | 0.0170 | 0.89 | 1.11 (0.27–4.48) |
| H6 | A | T | T | 0.0358 | 0.0148 | 0.27 | 0.52 (0.16–1.64) |
| Global haplotype association p-value: 0.83 | |||||||
| SNP | rs12524494 (A > G) | rs636933 (G > A) | rs648595 (G > T) | rs761142 C > A | rs606548 (C > T) | rs17883901 (G > A) |
|---|---|---|---|---|---|---|
| rs12524494 (A > G) | −0.0073 | 0.0186 | 0.0235 | 0.0285 | −0.0018 | |
| 0.0091 | −0.0331 | 0.0427 | 0.0511 | 0.0020 | ||
| 0.0259 | −0.0653 | 0.0948 | 0.1103 | 0.0070 | ||
| rs636933 (G > A) | 0.1190 | 0.1358 | −0.0060 | 0.0133 | ||
| 0.1190 | −0.1280 | 0.0110 | 0.0090 | |||
| 0.0445 | −0.0852 | 0.0355 | 0.0130 | |||
| rs648595 (G > T) | D | 0.1360 | 0.0192 | 0.0110 | ||
| −0.1453 | −0.0321 | 0.0135 | ||||
| −0.1458 | −0.0817 | 0.0039 | ||||
| rs761142 C > A | 0.0254 | 0.0136 | ||||
| 0.0427 | −0.0095 | |||||
| 0.1153 | −0.0044 | |||||
| rs606548 (C > T) | −0.0025 | |||||
| 0.0020 | ||||||
| 0.0121 | ||||||
| SNP | rs12524494 | rs636933 | rs648595 | rs761142 | rs606548 | rs17883901 |
| rs12524494 (A > G) | 0.9907 | 0.8955 | 0.8603 | 0.8862 | 0.6553 | |
| 0.7522 | 1.0000 | 0.9773 | 0.9114 | 0.4012 | ||
| 0.9701 | 0.8450 | 0.8361 | 0.8289 | 0.5638 | ||
| rs636933 (G > A) | 1.0000 | 0.8640 | 0.8669 | 0.2151 | ||
| 1.0000 | 0.8655 | 0.9174 | 0.1350 | |||
| 1.0000 | 0.9008 | 0.9269 | 0.2219 | |||
| rs648595 (G > T) | D’ | 0.9888 | 0.9967 | 0.2439 | ||
| 0.9799 | 0.9700 | 0.2917 | ||||
| 0.9206 | 0.7383 | 0.1301 | ||||
| rs761142 C > A | 0.9975 | 0.2291 | ||||
| 0.9773 | 0.1550 | |||||
| 0.7108 | 0.1011 | |||||
| rs606548 (C > T) | 0.9698 | |||||
| 0.4012 | ||||||
| 0.6841 | ||||||
| SNP | rs7517826 (C > A) | rs3827715 (T > C) | rs2301022 (C > T) |
|---|---|---|---|
| rs7517826 (C > A) | D | 0.1699 | −0.0730 |
| 0.1578 | 0.0778 | ||
| 0.1289 | −0.0035 | ||
| rs3827715 (T > C) | −0.0587 | ||
| 0.0661 | |||
| 0.0487 | |||
| SNP | rs7517826 | rs3827715 | rs2301022 |
| rs7517826 (C > A) | D’ | 0.9930 | 0.6658 |
| 0.9937 | 0.6509 | ||
| 0.9985 | 0.0155 | ||
| rs3827715 (T > C) | 0.7374 | ||
| 0.7778 | |||
| 0.5675 |
| Gene, Effective Allele | Stroke Phenotype | p-Value | Beta/Odds Ratio | Dataset | Sample Size |
|---|---|---|---|---|---|
| GCLC rs12524494-G | TOAST large artery atherosclerosis | 0.006 | ▲ 2.9874 | MEGASTROKE GWAS | 230, 076 |
| 0.049 | ▲ 3.0648 | MEGASTROKE GWAS (EUR) | 190, 513 | ||
| 0.26 | ▲ 1.4612 | CADISP 2015 | 9, 326 | ||
| 0.92 | ▼ 0.9674 | VHIR FMT 2018 | 783 | ||
| All ischemic stroke | 0.38 | ▲ 2.7532 | MEGASTROKE GWAS | 481, 992 | |
| 0.13 | ▲ 2.8174 | MEGASTROKE GWAS (EUR) | 404, 881 | ||
| 0.09 | ▲ 1.2628 | CADISP 2015 | 9, 814 | ||
| 0.70 | ▼ 0.9140 | VHIR FMT 2018 | 783 | ||
| Transient cerebral ischemic attacks and related syndromes | 0.136 | ▲ 1.10 | UK BIOBANK | 452, 264 | |
| Stroke, not specified as hemorrhage or infarction | 0.0017 | ▼ 0.745 | UK BIOBANK | 452, 264 | |
| GCLC rs17883901-A | TOAST large artery atherosclerosis | 0.16 | ▲ 2.8613 | MEGASTROKE GWAS | 227, 794 |
| 0.23 | ▲ 2.8871 | MEGASTROKE GWAS (EUR) | 192, 425 | ||
| 0.08 | ▲ 1.7191 | CADISP 2015 | 9, 326 | ||
| 0.06 | ▲ 2.1453 | VHIR FMT 2018 | 783 | ||
| All ischemic stroke | 0.09 | ▲ 2.7946 | MEGASTROKE GWAS | 475, 907 | |
| 0.74 | ▲ 2.7366 | MEGASTROKE GWAS (EUR) | 403, 224 | ||
| 0.23 | ▲ 1.1652 | CADISP 2015 | 9, 814 | ||
| 0.023 | ▲ 1.6958 | VHIR FMT 2018 | 783 | ||
| Transient cerebral ischemic attacks and related syndromes | 0.25 | ▲ 1.06 (G) | UK BIOBANK | 452, 264 | |
| Stroke, not specified as hemorrhage or infarction | 0.09 | ▼ 0.882 (G) | UK BIOBANK | 452, 264 | |
| GCLC rs606548-T | TOAST large artery atherosclerosis | 0.055 | ▲ 2.8984 | MEGASTROKE GWAS | 229, 842 |
| 0.036 | ▲ 3.1030 | MEGASTROKE GWAS (EUR) | 189, 632 | ||
| 0.10 | ▲ 1.7444 | CADISP 2015 | 9, 326 | ||
| 0.39 | ▼ 0.7489 | VHIR FMT 2018 | 783 | ||
| All ischemic stroke | 0.35 | ▲ 2.7541 | MEGASTROKE GWAS | 472, 735 | |
| 0.012 | ▲ 2.8929 | MEGASTROKE GWAS (EUR) | 395, 530 | ||
| 0.039 | ▲ 1.3340 | CADISP 2015 | 9, 814 | ||
| 0.42 | ▼ 0.8337 | VHIR FMT 2018 | 783 | ||
| Transient cerebral ischemic attacks and related syndromes | 0.34 | ▲ 1.06 | UK BIOBANK | 452, 264 | |
| Stroke, not specified as hemorrhage or infarction | 0.002 | ▼ 0.746 | UK BIOBANK | 452, 264 | |
| * GCLC rs636933-A | TOAST large artery atherosclerosis | 0.31 | ▲ 2.7857 | MEGASTROKE GWAS | 241, 607 |
| 0.37 | ▲ 2.7900 | MEGASTROKE GWAS (EUR) | 203, 144 | ||
| 0.33 | ▼ 0.8116 | CADISP 2015 | 9, 326 | ||
| 0.89 | ▲ 1.0272 | VHIR FMT 2018 | 783 | ||
| All ischemic stroke | 0.70 | ▲ 2.7077 | MEGASTROKE GWAS | 509, 234 | |
| 0.38 | ▲ 2.6911 | MEGASTROKE GWAS (EUR) | 432, 044 | ||
| 0.29 | ▼ 0.9216 | CADISP 2015 | 9, 814 | ||
| 0.24 | ▲ 1.1540 | VHIR FMT 2018 | 783 | ||
| Transient cerebral ischemic attacks and related syndromes | 0.53 | ▲ 1.02 (G) | UK BIOBANK | 452, 264 | |
| Stroke, not specified as hemorrhage or infarction | 0.51 | ▼ 0.97 (G) | UK BIOBANK | 452, 264 | |
| * GCLC rs648595-T | TOAST large artery atherosclerosis | 0.06 | ▲ 2.8283 | MEGASTROKE GWAS | 241, 442 |
| 0.23 | ▲ 2.8026 | MEGASTROKE GWAS (EUR) | 201, 232 | ||
| 0.61 | ▲ 1.0952 | CADISP 2015 | 9, 326 | ||
| 0.08 | ▼ 0.7558 | VHIR FMT 2018 | 783 | ||
| All ischemic stroke | 0.46 | ▲ 2.7358 | MEGASTROKE GWAS | 500, 913 | |
| 0.33 | ▲ 2.7455 | MEGASTROKE GWAS (EUR) | 423, 708 | ||
| 0.93 | ▲ 1.0056 | CADISP 2015 | 9, 814 | ||
| 0.73 | ▲ 1.0362 | VHIR FMT 2018 | 783 | ||
| Transient cerebral ischemic attacks and related syndromes | 0.93 | ▲ 1.00 (G) | UK BIOBANK | 452, 264 | |
| Stroke, not specified as hemorrhage or infarction | 0.80 | ▲ 1.01 (G) | UK BIOBANK | 452, 264 | |
| * GCLC rs761142-C | TOAST large artery atherosclerosis | 0.04 | ▲ 2.8442 | MEGASTROKE GWAS | 240, 561 |
| 0.14 | ▲ 2.8359 | MEGASTROKE GWAS (EUR) | 200, 351 | ||
| 0.77 | ▼ 0.9415 | CADISP 2015 | 9, 326 | ||
| 0.78 | ▼ 0.9510 | VHIR FMT 2018 | 783 | ||
| All ischemic stroke | 0.63 | ▲ 2.7064 | MEGASTROKE GWAS | 499, 208 | |
| 0.65 | ▲ 2.7042 | MEGASTROKE GWAS (EUR) | 422, 020 | ||
| 0.89 | ▼ 0.9899 | CADISP 2015 | 9, 814 | ||
| 0.27 | ▲ 1.1348 | VHIR FMT 2018 | 783 | ||
| Transient cerebral ischemic attacks and related syndromes | 0.89 | ▲ 1.00 | UK BIOBANK | 452, 264 | |
| Stroke, not specified as hemorrhage or infarction | 0.48 | ▼ 0.97 | UK BIOBANK | 452, 264 | |
| * GCLM rs2301022-T | TOAST large artery atherosclerosis | 0.99 | ▲ 2.7172 | MEGASTROKE GWAS | 242, 987 |
| 0.74 | ▲ 2.6948 | MEGASTROKE GWAS (EUR) | 203, 144 | ||
| 0.96 | ▲ 1.0106 | CADISP 2015 | 9, 326 | ||
| 0.03 | ▲ 1.4255 | VHIR FMT 2018 | 783 | ||
| All ischemic stroke | 0.73 | ▲ 2.7099 | MEGASTROKE GWAS | 511, 623 | |
| 0.19 | ▲ 2.6808 | MEGASTROKE GWAS (EUR) | 434, 418 | ||
| 0.07 | ▲ 1.1338 | CADISP 2015 | 9, 814 | ||
| 0.07 | ▲ 1.2157 | VHIR FMT 2018 | 783 | ||
| Transient cerebral ischemic attacks and related syndromes | 0.08 | ▲ 1.05 (C) | UK BIOBANK | 452, 264 | |
| Stroke, not specified as hemorrhage or infarction | 0.12 | ▼ 0.93 (C) | UK BIOBANK | 452, 264 | |
| GCLM rs3827715-C | TOAST large artery atherosclerosis | 0.49 | ▲ 2.7639 | MEGASTROKE GWAS | 242, 987 |
| 0.74 | ▲ 2.7441 | MEGASTROKE GWAS (EUR) | 203, 144 | ||
| 0.79 | ▲ 1.0540 | CADISP 2015 | 9, 326 | ||
| 0.15 | ▲ 1.2955 | VHIR FMT 2018 | 783 | ||
| All ischemic stroke | 0.55 | ▲ 2.7341 | MEGASTROKE GWAS | 511, 561 | |
| 0.97 | ▲ 2.7169 | MEGASTROKE GWAS (EUR) | 434, 418 | ||
| 0.45 | ▲ 1.0572 | CADISP 2015 | 9, 814 | ||
| 0.02 | ▲ 1.3013 | VHIR FMT 2018 | 783 | ||
| Transient cerebral ischemic attacks and related syndromes | 0.26 | ▲ 1.03 | UK BIOBANK | 452, 264 | |
| Stroke, not specified as hemorrhage or infarction | 0.32 | ▲ 1.05 | UK BIOBANK | 452, 264 | |
| GCLM rs7517826-A | TOAST large artery atherosclerosis | 0.88 | ▲ 2.7091 | MEGASTROKE GWAS | 241, 442 |
| 0.45 | ▲ 2.6655 | MEGASTROKE GWAS (EUR) | 201, 232 | ||
| 0.65 | ▲ 1.0871 | CADISP 2015 | 9, 326 | ||
| 0.06 | ▲ 1.3540 | VHIR FMT 2018 | 783 | ||
| All ischemic stroke | 0.59 | ▲ 2.7053 | MEGASTROKE GWAS | 503, 288 | |
| 0.61 | ▲ 2.7040 | MEGASTROKE GWAS (EUR) | 426, 083 | ||
| 0.98 | ▼ 0.9982 | CADISP 2015 | 9, 814 | ||
| 0.026 | ▲ 1.2603 | VHIR FMT 2018 | 783 | ||
| Transient cerebral ischemic attacks and related syndromes | 0.019 | ▲ 1.07 | UK BIOBANK | 452, 264 | |
| Stroke, not specified as hemorrhage or infarction | 0.67 | ▲ 1.02 | UK BIOBANK | 452, 264 |
| Gene–Gene and Gene–Environment Interactions | NH | β H | WH | NL | β L | WL | Pperm | |
|---|---|---|---|---|---|---|---|---|
| Two-order GxG/GxE interactions | ||||||||
| 1 | GCLM rs2301022 × RASEF rs4322086 | 3 | 0.175 | 37.70 | 4 | −0.176 | 29.71 | <0.001 |
| 2 | SMOKE × ALCOHOL | 2 | 0.175 | 30.54 | 1 | −0.176 | 32.33 | <0.001 |
| 3 | SMOKE × VEGET | 2 | 0.176 | 30.79 | 1 | −0.147 | 20.61 | <0.001 |
| 4 | RASEF rs4322086 × SMOKE | 1 | 0.199 | 30.24 | 1 | −0.141 | 16.50 | <0.001 |
| Three-order GxG/GxE interactions | ||||||||
| 1 | GCLM rs2301022 × RASEF rs4322086 × LOC105370913 rs899997 | 3 | 0.173 | 29.22 | 6 | −0.248 | 45.37 | <0.001 |
| 2 | GCLM rs3827715 × GCLM rs2301022 × RASEF rs4322086 | 3 | 0.143 | 17.27 | 5 | −0.240 | 45.12 | <0.001 |
| 3 | GCLC rs648595 × RASEF rs4322086 × ZC3HC1 rs11556924 | 4 | 0.234 | 43.84 | 1 | −0.170 | 8.47 | <0.001 |
| 4 | GCLM rs2301022 × LDLR rs6511720 × RASEF rs4322086 | 5 | 0.195 | 43.54 | 4 | −0.195 | 31.64 | <0.001 |
| Four-order GxG/GxE interactions | ||||||||
| 1 | GCLM rs2301022 × RASEF rs4322086 × LOC105370913 rs899997 × SMOKE | 6 | 0.229 | 36.43 | 9 | −0.273 | 59.01 | <0.002 |
| 2 | GCLM rs3827715 × GCLM rs2301022 × GCLC rs761142 × RASEF rs4322086 | 7 | 0.252 | 46.33 | 10 | −0.260 | 55.87 | <0.002 |
| 3 | GCLM rs2301022 × GCLC rs606548 × RASEF rs4322086 × SMOKE | 6 | 0.257 | 55.05 | 5 | −0.180 | 26.83 | <0.002 |
| 4 | GCLC rs648595 × LDLR rs6511720 × RASEF rs4322086 × ZC3HC1 rs11556924 | 6 | 0.272 | 54.15 | 5 | −0.219 | 24.02 | <0.002 |
| Five-order GxG/GxE interactions | ||||||||
| 1 | GCLM rs3827715 × GCLC rs17883901 × GCLC rs12524494 ALCOHOL × SMOKE | 1 | 0.093 | 3.43 | 5 | −0.139 | 19.42 | 0.01 |
| 2 | GCLC rs636933 × RASEF rs4322086 × SLCO1B1 rs2417957 × PITX2 rs12646447 × VEGET | 1 | 0.159 | 8.08 | 3 | −0.237 | 19.37 | 0.01 |
| 3 | GCLM rs2301022 × GCLC rs12524494 × AIM1 rs783396 × SLCO1B1 rs2417957 × VEGET | 3 | 0.142 | 12.34 | 4 | −0.155 | 19.36 | 0.01 |
| 4 | GCLM rs7517826 × GCLC rs606548 × GCLC rs12524494 × PEMT rs12449964 × VEGET | 1 | 0.237 | 4.65 | 7 | −0.215 | 19.35 | 0.01 |
| № | Genotype Combinations | IS Patients | Controls | OR (95% CI) 1 | p 2 | FDR 3 | ||
|---|---|---|---|---|---|---|---|---|
| n | % | n | % | |||||
| 1 | RASEF rs4322086-G/G × GCLM rs2301022-C/C | 43 | 7.9 | 56 | 8.7 | 0.89 (0.59–1.35) | 0.599 | 0.63 |
| 2 | RASEF rs4322086-G/G × GCLM rs2301022-C/T | 45 | 8.3 | 31 | 4.8 | 1.77 (1.10–2.84) | 0.017 | 0.04 |
| 3 | RASEF rs4322086-G/G × GCLM rs2301022-T/T | 4 | 0.7 | 16 | 2.5 | 0.29 (0.10–0.87) | 0.019 | 0.04 |
| 4 | RASEF rs4322086-G/A × GCLM rs2301022-C/C | 157 | 28.8 | 137 | 21.4 | 1.49 (1.14–1.94) | 0.003 | 0.015 |
| 5 | RASEF rs4322086-G/A × GCLM rs2301022-C/T | 127 | 23.3 | 106 | 16.5 | 1.53 (1.15–2.04) | 0.003 | 0.015 |
| 6 | RASEF rs4322086-G/A × GCLM rs2301022-T/T | 14 | 2.6 | 38 | 5.9 | 0.42 (0.22–0.78) | 0.005 | 0.015 |
| 7 | RASEF rs4322086-A/A × GCLM rs2301022-C/C | 80 | 14.7 | 135 | 21.1 | 0.64 (0.48–0.87) | 0.004 | 0.015 |
| 8 | RASEF rs4322086-A/A × GCLM rs2301022-C/T | 67 | 12.3 | 97 | 15.1 | 0.79 (0.56–1.10) | 0.158 | 0.26 |
| 9 | RASEF rs4322086-A/A × GCLM rs2301022-T/T | 8 | 1.5 | 25 | 3.9 | 0.38 (0.17–0.84) | 0.020 | 0.04 |
| 10 | PEMT rs12449964-C/C × GCLM rs2301022-C/C | 109 | 20.1 | 124 | 18.7 | 1.10 (0.82–1.46) | 0.528 | 0.59 |
| 11 | PEMT rs12449964-C/C × GCLM rs2301022-C/T | 92 | 17.0 | 87 | 13.1 | 1.36 (0.99–1.86) | 0.060 | 0.11 |
| 12 | PEMT rs12449964-C/C × GCLM rs2301022-T/T | 11 | 2.0 | 34 | 5.1 | 0.38 (0.19–0.77) | 0.005 | 0.015 |
| 13 | PEMT rs12449964-C/T × GCLM rs2301022-C/C | 139 | 25.7 | 164 | 24.7 | 1.05 (0.81–1.37) | 0.703 | 0.70 |
| 14 | PEMT rs12449964-C/T × GCLM rs2301022-C/T | 113 | 20.9 | 125 | 18.9 | 1.14 (0.86–1.51) | 0.378 | 0.49 |
| 15 | PEMT rs12449964-C/T × GCLM rs2301022-T/T | 9 | 1.7 | 42 | 6.3 | 0.26 (0.13–0.53) | 0.0001 | 0.0018 |
| 16 | PEMT rs12449964-T/T × GCLM rs2301022-C/C | 32 | 5.9 | 52 | 7.8 | 0.74 (0.47–1.17) | 0.191 | 0.29 |
| 17 | PEMT rs12449964-T/T × GCLM rs2301022-C/T | 30 | 5.5 | 31 | 4.7 | 1.20 (0.71–2.00) | 0.494 | 0.59 |
| 18 | PEMT rs12449964-T/T × GCLM rs2301022-T/T | 6 | 1.1 | 4 | 0.6 | 1.78 (0.53–5.95) | 0.336 | 0.47 |
| Gene–Gene and Gene–Environment Interactions | NH | β H | WH | NL | β L | WL | Pperm | |
|---|---|---|---|---|---|---|---|---|
| Two-order GxG/GxE interactions | ||||||||
| 1 | RASEF rs4322086 × SMOKE | 1 | 3.968 | 13.93 | 1 | −2.375 | 5.41 | 0.002 |
| 2 | RASEF rs4322086 × ZC3HC1 rs11556924 | 2 | 3.863 | 16.19 | 2 | −2.952 | 8.11 | 0.005 |
| 3 | RASEF rs4322086 × GCLM rs2301022 | 2 | 3.239 | 14.65 | 2 | −2.589 | 5.99 | 0.008 |
| 4 | SLCO1B1 rs2417957 × GCLC rs648595 | 2 | 21.281 | 19.80 | 0 | - | - | 0.009 |
| Three-order GxG/GxE interactions | ||||||||
| 1 | PITX2 rs12646447 × ZC3HC1 rs11556924 × GCLC rs648595 | 3 | 27.429 | 38.68 | 0 | - | - | 0.001 |
| 2 | ZC3HC1 rs11556924 × GCLC rs648595 × ALCOHOL | 3 | 9.974 | 26.38 | 0 | - | - | 0.001 |
| 3 | PITX2 rs12646447 × PEMT rs12449964 × GCLC rs648595 | 2 | 44.033 | 30.18 | 0 | - | - | 0.002 |
| 4 | RASEF rs4322086 × PITX2 rs12646447 × SMOKE | 4 | 6.681 | 32.73 | 2 | −3.749 | 9.84 | 0.004 |
| Four-order GxG/GxE interactions | ||||||||
| 1 | GCLM rs3827715 × GCLC rs636933 × PITX2 rs12646447 × ZC3HC1 rs11556924 | 4 | 28.155 | 71.44 | 0 | - | - | <0.002 |
| 2 | GCLC rs648595 × SLCO1B1 rs2417957 × PITX2 rs12646447 × ZC3HC1 rs11556924 | 5 | 28.427 | 56.95 | 1 | −7.303 | 2.77 | <0.002 |
| 3 | GCLC rs648595 × GCLC rs606548 × PITX2 rs12646447 × ZC3HC1 rs11556924 | 5 | 21.454 | 54.06 | 0 | - | - | <0.002 |
| 4 | GCLC rs648595 × PEMT rs12449964 × ZC3HC1 rs11556924 × ALCOHOL | 5 | 17.308 | 53.83 | 0 | - | - | <0.002 |
| Five-order GxG/GxE interactions | ||||||||
| 1 | GCLC rs636933 × GCLC rs12524494 × AIM1 rs783396 × SLCO1B1 rs2417957 × PITX2 rs12646447 | 4 | 14.795 | 18.69 | 1 | −3.124 | 2.74 | 0.01 |
| 2 | GCLC rs606548 × GCLC rs648595 × GCLC rs17883901 × SLCO1B1 rs2417957 × PITX2 rs12646447 | 6 | 7.699 | 18.65 | 1 | −6.380 | 3.41 | 0.01 |
| 3 | GCLC rs606548 × SLCO1B1 rs2417957 × ZC3HC1 rs11556924 × VEGET × ALCOHOL | 3 | 35.186 | 18.23 | 1 | −2.748 | 2.81 | 0.01 |
| 4 | GCLC rs606548 × GCLC rs12524494 × GCLC rs17883901 × PEMT rs12449964 × ALCOHOL | 4 | 10.448 | 18.10 | 1 | −3.016 | 4.53 | 0.01 |
| Gene | SNP ID | Alleles | Location in the Gene | Regulatory Potential | Expression Levels (eQTL Analysis) | Epigenetic Regulation | TFBS | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FuncPred | Regulome Score | Blood/ Blood Cells | Arteries, Aorta | Brain Tissues | Histone Marks | Open Chromatin | CTCF Binding | Promoter | Promoter Flanking region | DNA Methylation | VEP | Transfac | atSNP | ||||||||||
| GTEx | eQTLGen | QTLbase | GTEx | QTLbase | GTEx | QTLbase | Blood | Arteries, Aorta | Brain Tissues | ||||||||||||||
| GCLC | rs12524494 | A/G | intron | 0.000 | 6 | √ | √ | √ | √ | ||||||||||||||
| rs17883901 | G/A | intron | 0.249 | 3a | √ | √ | √ | √ | √ | √ | √ | √ | |||||||||||
| rs606548 | C/T | intron | 0.000 | 5 | √ | √ | √ | √ | |||||||||||||||
| rs636933 | G/A | intron | - | - | √ | √ | √ | √ | |||||||||||||||
| rs648595 | G/T | intron | 0.187 | 5 | √ | √ | √ | √ | √ | ||||||||||||||
| rs761142 | A/C | intron | 0.000 | 5 | √ | √ | √ | √ | √ | √ | √ | ||||||||||||
| GCLM | rs2301022 | C/T | intron | 0.000 | 4 | √ | √ | √ | √ | √ | √ | √ | |||||||||||
| rs3827715 | T/C | intron | 0.000 | 5 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | |||||||||
| rs7517826 | C/A | intron | 0.000 | - | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polonikov, A.; Bocharova, I.; Azarova, I.; Klyosova, E.; Bykanova, M.; Bushueva, O.; Polonikova, A.; Churnosov, M.; Solodilova, M. The Impact of Genetic Polymorphisms in Glutamate-Cysteine Ligase, a Key Enzyme of Glutathione Biosynthesis, on Ischemic Stroke Risk and Brain Infarct Size. Life 2022, 12, 602. https://doi.org/10.3390/life12040602
Polonikov A, Bocharova I, Azarova I, Klyosova E, Bykanova M, Bushueva O, Polonikova A, Churnosov M, Solodilova M. The Impact of Genetic Polymorphisms in Glutamate-Cysteine Ligase, a Key Enzyme of Glutathione Biosynthesis, on Ischemic Stroke Risk and Brain Infarct Size. Life. 2022; 12(4):602. https://doi.org/10.3390/life12040602
Chicago/Turabian StylePolonikov, Alexey, Iuliia Bocharova, Iuliia Azarova, Elena Klyosova, Marina Bykanova, Olga Bushueva, Anna Polonikova, Mikhail Churnosov, and Maria Solodilova. 2022. "The Impact of Genetic Polymorphisms in Glutamate-Cysteine Ligase, a Key Enzyme of Glutathione Biosynthesis, on Ischemic Stroke Risk and Brain Infarct Size" Life 12, no. 4: 602. https://doi.org/10.3390/life12040602
APA StylePolonikov, A., Bocharova, I., Azarova, I., Klyosova, E., Bykanova, M., Bushueva, O., Polonikova, A., Churnosov, M., & Solodilova, M. (2022). The Impact of Genetic Polymorphisms in Glutamate-Cysteine Ligase, a Key Enzyme of Glutathione Biosynthesis, on Ischemic Stroke Risk and Brain Infarct Size. Life, 12(4), 602. https://doi.org/10.3390/life12040602