
Dopamine and Dopamine-Related Ligands Can Bind Not Only to Dopamine Receptors


Abstract
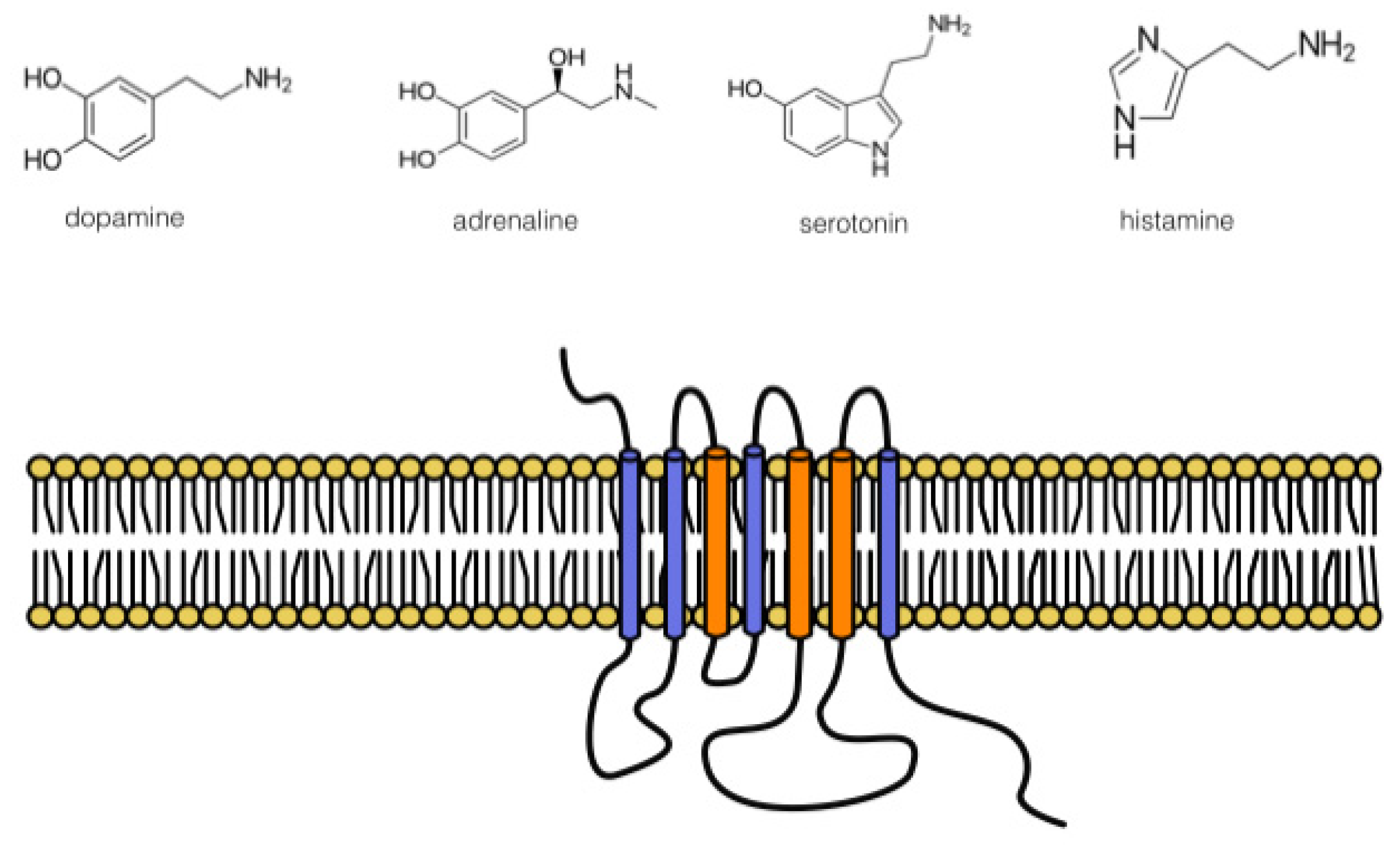
:1. Introduction
1.1. D1-like Family
1.2. D2-like Family
1.3. DR Ligand Targets
2. DR Agonists
2.1. So-Called Selective Dopamine Receptor Agonists
2.2. Drugs–Dopamine Receptor Agonists with Multiple Targets of Action
3. DR Antagonists
3.1. So-Called Selective Dopamine Receptor Antagonists
3.2. Drugs–Dopamine Receptor Antagonists with Multiple Targets of Action
4. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Explanation |
| DR(s) | Dopamine receptor(s) |
| ARs | Adrenoceptors |
| 5-HT | Serotonin |
| TM | Transmembrane zone |
| pKi | The negative logarithm of the Ki value (the molar concentration of the competing ligand that would occupy 50% of the receptors) |
| pKD | The negative logarithm of KD value (the equilibrium dissociation constant represents the concentration of radioligand occupying half of the maximum receptor population) |
| pA2 | The measure of the potency of an antagonist, negative logarithm of the molar concentration of an antagonist that would produce a two-fold shift in the concentration-response curve for an agonist |
| pEC50 | The negative logarithm of EC50 value (the molar concentration of an agonist that produces 50% of the maximum possible response for that agonist). This value can vary when comparing different activation pathways |
References
- Emilien, G.; Maloteaux, J.-M.; Geurts, M.; Hoogenberg, K.; Cragg, S. Dopamine receptors—Physiological understanding to therapeutic intervention potential. Pharmacol. Ther. 1999, 84, 133–156. [Google Scholar] [CrossRef]
- Undieh, A.S. Pharmacology of signaling induced by dopamine D1-like receptor activation. Pharmacol. Ther. 2010, 128, 37–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Civelli, O.; Bunzow, J.K.; Grandy, D.K. Molecular Diversity of the Dopamine Receptors. Annu. Rev. Pharmacol. Toxicol. 1993, 33, 281–307. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, B.F. Structures of Dopamine Receptors. J. Neurochem. 1993, 60, 804–816. [Google Scholar] [CrossRef]
- Giros, B.; Sokoloff, P.; Martres, M.P.; Riou, J.F.; Emorine, L.J.; Schwartz, J.C. Alternative splicing directs the expression of two D2 dopamine receptor isoforms. Nature 1989, 342, 923–926. [Google Scholar] [CrossRef]
- Grandy, D.K.; Litt, M.; Allen, L.; Bunzow, J.R.; Marchionni, M.; Makam, H.; Reed, L.; Magenis, R.E.; Civelli, O. The human dopamine D2 receptor gene is located on chromosome 11 at q22–q23 and identifies a TaqI RFLP. Am. J. Hum. Genet. 1989, 45, 778–785. [Google Scholar]
- Sokoloff, P.; Giros, B.; Martres, M.P.; Bouthenet, M.L.; Schwartz, J.C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146–151. [Google Scholar] [CrossRef]
- Van Tol, H.H.; Bunzow, J.R.; Guan, H.C.; Sunahara, R.K.; Seeman, P.; Niznik, H.B.; Civelli, O. Cloning of the gene for a human dopamine D4 receptor with high affinity for the antipsychotic clozapine. Nature 1991, 350, 610–614. [Google Scholar] [CrossRef]
- Liu, L.X.; Monsma, F.J., Jr.; Sibley, D.R.; Chiodo, L.A. D2L, D2S, and D3 dopamine receptors stably transfected into NG108-15 cells couple to a voltage-dependent potassium current via distinct G protein mechanisms. Synapse 1996, 24, 156–164. [Google Scholar] [CrossRef]
- Picetti, R.; Saiardi, A.; Abdel Samad, T.; Bozzi, Y.; Baik, J.H.; Borrelli, E. Dopamine D2 receptors in signal transduction and behavior. Crit. Rev. Neurobiol. 1997, 11, 121–142. [Google Scholar] [CrossRef]
- Weinshank, R.L.; Adham, N.; Macchi, M.; Olsen, M.A.; Branchek, T.A.; Hartig, P.R. Molecular cloning and characterization of a high affinity dopamine receptor (D1 beta) and its pseudogene. J. Biol. Chem. 1991, 266, 22427–22435. [Google Scholar] [CrossRef]
- Centonze, D.; Grande, C.; Saulle, E.; Martin, A.B.; Gubellini, P.; Pavón, N.; Pisani, A.; Bernardi, G.; Moratalla, R.; Calabresi, P. Distinct roles of D1 and D5 dopamine receptors in motor activity and striatal synaptic plasticity. J. Neurosci. 2003, 23, 8506–8512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiberi, M.; Caron, M.G. High agonist-independent activity is a distinguishing feature of the dopamine D1B receptor subtype. J. Biol. Chem. 1994, 269, 27925–27931. [Google Scholar] [CrossRef]
- Wang, Q.; Jolly, J.P.; Surmeier, J.D.; Mullah, B.M.; Lidow, M.S.; Bergson, C.M.; Robishaw, J.D. Differential dependence of the D1 and D5 dopamine receptors on the G protein gamma 7 subunit for activation of adenylylcyclase. J. Biol. Chem. 2001, 276, 39386–39393. [Google Scholar] [CrossRef] [Green Version]
- Herve, D.; Levi-Strauss, M.; Marey-Semper, I.; Verney, C.; Tassin, J.P.; Glowinski, J.; Girault, J.A. G(olf) and Gs in rat basal ganglia: Possible involvement of G(olf) in the coupling of dopamine D1 receptor with adenylyl cyclase. J. Neurosci. 1993, 13, 2237–2248. [Google Scholar] [CrossRef] [Green Version]
- Hollon, T.R.; Bek, M.J.; Lachowicz, J.E.; Ariano, M.A.; Mezey, E.; Ramachandran, R.; Wersinger, S.R.; Soares-da-Silva, P.; Liu, Z.F.; Grinberg, A.; et al. Mice lacking D5 dopamine receptors have increased sympathetic tone and are hypertensive. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 10801–10810. [Google Scholar] [CrossRef] [Green Version]
- Weiner, D.M.; Levey, A.I.; Sunahara, R.K.; Niznik, H.B.; O´Dowd, B.F.; Seeman, P.; Brann, M.R. D1 and D2 dopamine receptor mRNA in rat brain. Proc. Natl. Acad. Sci. USA 1991, 88, 1859–1863. [Google Scholar] [CrossRef] [Green Version]
- Khan, Z.U.; Mrzljak, L.; Gutierrez, A.; de la Calle, A.; Goldman-Rakic, P.S. Prominence of the dopamine D2 short isoform in dopaminergic pathways. Proc. Natl. Acad. Sci. USA 1998, 95, 7731–7736. [Google Scholar] [CrossRef] [Green Version]
- Saiardi, A.; Abdel Samad, T.; Picetti, R.; Bozzi, Y.; Baik, J.H.; Borrelli, E. The physiological role of dopamine D2 receptors. Adv. Pharmacol. 1998, 42, 521–524. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Levesque, D.; Martres, M.P.; Sokoloff, P. Dopamine D3 receptor: Basic and clinical aspects. Clin. Neuropharmacol. 1993, 16, 295–314. [Google Scholar] [CrossRef]
- Dziedzicka-Wasylewska, M. Brain dopamine receptors—Research perspectives and potential sites of regulation. Pol. J. Pharmacol. 2004, 56, 659–671. [Google Scholar] [PubMed]
- Patel, S.; Patel, S.; Marwood, R.; Emms, F.; Marston, D.; Leeson, P.D.; Curtis, N.R.; Kulagowski, J.J.; Freedman, S.B. Identification and pharmacological characterization of [125I]L-750,667, a novel radioligand for the dopamine D4 receptor. Mol. Pharmacol. 1996, 50, 1658–1664. [Google Scholar] [PubMed]
- Sunahara, R.K.; Guan, H.C.; O´Dowd, B.F.; Seeman, P.; Laurier, L.G.; Ng, G.; George, S.R.; Torchia, J.; Van Tol, H.H.; Niznik, H.B. Cloning of the gene for a human dopamine D5 receptor with higher affinity for dopamine than D1. Nature 1991, 350, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.M.; Kung, M.-P.; Kabalka, G.W.; Kung, H.F.; Switzer, R. Synthesis and Characterization of Radioiodinated N-(3-Iodopropen-1-yl)-2 β-carbomethoxy-3 β-(4-chlorophenyl)tropanes: Potential Dopamine Reuptake Site Imaging Agents. J. Med. Chem. 1994, 37, 1535–1542. [Google Scholar] [CrossRef]
- Meltzer, P.C.; McPhee, M.; Madras, B.K. Synthesis and biological activity of 2-Carbomethoxy-3-catechol-8-azabicyclo[3.2.1]octanes. Bioorg. Med. Chem. Lett. 2003, 13, 4133–4137. [Google Scholar] [CrossRef]
- Fisher, L.E.; Rosenkranz, R.P.; Clark, R.D.; Muchowski, J.M.; McClelland, D.L.; Michel, A.; Caroon, J.M.; Galeazzi, E.; Eglen, R.; Whiting, R.L. N,N-6-bis-[2-(3,4-dihydroxybenzyl)pyrrolidinyl]hexane, a potent, selective, orally active dopamine analog with hypotensive and diuretic activity. Bioorg. Med. Chem. Lett. 1995, 5, 2371–2376. [Google Scholar] [CrossRef]
- Lu, S.-F.; Herbert, B.; Haufe, G.; Laue, K.W.; Padgett, W.L.; Oshunleti, O.; Daly, J.W.; Kirk, K.L. Syntheses of (R)-and (S)-2- and 6-Fluoronorepinephrine and (R)- and (S)-2- and 6-Fluoroepinephrine: Effect of Stereochemistry on Fluorine-Induced Adrenergic Selectivities. J. Med. Chem. 2000, 43, 1611–1619. [Google Scholar] [CrossRef]
- Kenakin, T. What is pharmacological ‘affinity’? Relevance to biased agonism and antagonism. Trends Pharmacol. Sci. 2014, 35, 434–441. [Google Scholar] [CrossRef]
- Rupniak, N.M.J.; Perdona, E.; Griffante, C.; Cavallini, P.; Sava, A.; Ricca, D.J.; Thor, K.B.; Burgard, E.C.; Corsi, M. Affinity, potency, efficacy, and selectivity of neurokinin A analogs at human recombinant NK2 and NK1 receptors. PLoS ONE 2018, 13, e0205894. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Cai, W.; Yu, L.; Zhen, X.; Zhang, A. ‘Click’ D1 receptor agonists with a 5-HT1A receptor pharmacophore producing D2 receptor activity. Bioorg. Med. Chem. 2009, 17, 4873–4880. [Google Scholar] [CrossRef]
- DeNinno, M.P.; Schoenleber, R.; Asin, K.E.; MacKenzie, R.; Kebabian, J.W. (1R,3S)-1-(Aminomethyl)-3,4-dihydro-5,6-dihydroxy-3-phenyl-1H-2-benzopyran: A potent and selective D1 agonist. J. Med. Chem. 1990, 33, 2948–2950. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Zhang, Y.; Yang, Z.; Qiang, K.; Chen, C.; Sun, L.; Chen, M.; Zhang, J. Chemical synthesis, microbial transformation and biological evaluation of tetrahydroprotoberberines as dopamine D1/D2 receptor ligands. Bioorg. Med. Chem. 2019, 27, 2100–2111. [Google Scholar] [CrossRef] [PubMed]
- Lebar, M.D.; Hahn, K.N.; Mutka, T.; Maignan, P.; McClintock, J.B.; Amsler, C.D.; van Olphen, A.; Kyle, D.E.; Baker, B.J. CNS and antimalarial activity of synthetic meridianin and psammopemmin analogs. Bioorg. Med. Chem. 2011, 19, 5756–5762. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Yan, W.; McCorvy, J.D.; Cheng, J. Biased Ligands of G Protein-Coupled Receptors (GPCRs): Structure-Functional Selectivity Relationships (SFSRs) and Therapeutic Potential. J. Med. Chem. 2018, 61, 9841–9878. [Google Scholar] [CrossRef] [PubMed]
- Kopinathan, A.; Scammells, P.J.; Lane, J.R.; Capuano, B. Multivalent approaches and beyond: Novel tools for the investigation of dopamine D2 receptor pharmacology. Future Med. Chem. 2016, 8, 1349–1372. [Google Scholar] [CrossRef] [PubMed]
- Żuk, J.; Bartuzi, D.; Miszta, P.; Kaczor, A.A. The Role of Lipids in Allosteric Modulation of Dopamine D(2) Receptor-In Silico Study. Molecules 2022, 27, 1335. [Google Scholar] [CrossRef]
- Jones-Tabah, J.; Mohammad, H.; Paulus, E.G.; Clarke, P.B.S.; Hébert, T.E. The Signaling and Pharmacology of the Dopamine D1 Receptor. Front. Cell. Neurosci. 2021, 15, 806618. [Google Scholar] [CrossRef]
- Free, R.B.; Chun, L.S.; Moritz, A.E.; Miller, B.N.; Doyle, T.B.; Conroy, J.L.; Padron, A.; Meade, J.A.; Xiao, J.; Hu, X.; et al. Discovery and characterization of a G protein-biased agonist that inhibits β-arrestin recruitment to the D2 dopamine receptor. Mol. Pharmacol. 2014, 86, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Bouthenet, M.L.; Ruat, M.; Sales, N.; Garbarg, M.; Schwartz, J.C. A detailed mapping of hist amine H1-receptors in guinea-pig central nervous system established by autoradiography with [125I]iodobolpyramine. Neuroscience 1988, 26, 553–600. [Google Scholar] [CrossRef]
- Vallone, D.; Picetti, R.; Borrelli, E. Structure and function of dopamine receptors. Neurosci. Biobehav. Rev. 2000, 24, 125–132. [Google Scholar] [CrossRef]
- Saunders, C.; Limbird, L.E. Localization and trafficking of α2-adrenergic receptor subtypes in cells and tissues. Pharmacol. Ther. 1999, 84, 193–205. [Google Scholar] [CrossRef]
- Nichols, D.E.; Nichols, C.D. Serotonin Receptors. Chem. Rev. 2008, 108, 1614–1641. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.J.; Smith, C.T.; Petersen, K.J.; Trujillo, P.; van Wouwe, N.C.; Donahue, M.J.; Kessler, R.M.; Deutch, A.Y.; Zald, D.H.; Claassen, D.O. [(18)F]fallypride characterization of striatal and extrastriatal D(2/3) receptors in Parkinson’s disease. Neuroimage Clin. 2018, 18, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Amenta, F.; Mignini, F.; Ricci, A.; Sabbatini, M.; Tomassoni, D.; Tayebati, S.K. Age-related changes of dopamine receptors in the rat hippocampus: A light microscope autoradiography study. Mech. Ageing Dev. 2001, 122, 2071–2083. [Google Scholar] [CrossRef]
- Szőllősi, E.; Bobok, A.; Kiss, L.; Vass, M.; Kurkó, D.; Kolok, S.; Visegrády, A.; Keserű, G.M. Cell-based and virtual fragment screening for adrenergic α2C receptor agonists. Bioorg. Med. Chem. 2015, 23, 3991–3999. [Google Scholar] [CrossRef] [Green Version]
- Nergårdh, R.; Oerther, S.; Fredholm, B.B. Differences between A 68930 and SKF 82958 could suggest synergistic roles of D1 and D5 receptors. Pharmacol. Biochem. Behav. 2005, 82, 495–505. [Google Scholar] [CrossRef]
- DeNinno, M.P.; Schoenleber, R.; Perner, R.J.; Lijewski, L.; Asin, K.E.; Britton, D.R.; MacKenzie, R.; Kebabian, J.W. Synthesis and dopaminergic activity of 3-substituted 1-(aminomethyl)-3,4-dihydro-5,6-dihydroxy-1H-2-benzopyrans: Characterization of an auxiliary binding region in the D1 receptor. J. Med. Chem. 1991, 34, 2561–2569. [Google Scholar] [CrossRef]
- Stuchlik, A.; Radostová, D.; Hatalova, H.; Vales, K.; Nekovarova, T.; Koprivova, J.; Svoboda, J.; Horacek, J. Validity of Quinpirole Sensitization Rat Model of OCD: Linking Evidence from Animal and Clinical Studies. Front. Behav. Neurosci. 2016, 10, 209. [Google Scholar] [CrossRef] [Green Version]
- Burris, K.D.; Pacheco, M.A.; Filtz, T.M.; Kung, M.P.; Kung, H.F.; Molinoff, P.B. Lack of discrimination by agonists for D2 and D3 dopamine receptors. Neuropsychopharmacology 1995, 12, 335–345. [Google Scholar] [CrossRef]
- Millan, M.J.; Maiofiss, L.; Cussac, D.; Audinot, V.; Boutin, J.A.; Newman-Tancredi, A. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes. J. Pharmacol. Exp. Ther. 2002, 303, 791–804. [Google Scholar] [CrossRef] [Green Version]
- Möller, D.; Kling, R.C.; Skultety, M.; Leuner, K.; Hübner, H.; Gmeiner, P. Functionally selective dopamine D2, D3 receptor partial agonists. J. Med. Chem. 2014, 57, 4861–4875. [Google Scholar] [CrossRef]
- Elsner, J.; Boeckler, F.; Heinemann, F.W.; Hübner, H.; Gmeiner, P. Pharmacophore-guided drug discovery investigations leading to bioactive 5-aminotetrahydropyrazolopyridines. Implications for the binding mode of heterocyclic dopamine D3 receptor agonists. J. Med. Chem. 2005, 48, 5771–5779. [Google Scholar] [CrossRef] [PubMed]
- Newman-Tancredi, A.; Cussac, D.; Audinot, V.; Millan, M.J. Actions of roxindole at recombinant human dopamine D2, D3 and D4 and serotonin 5-HT1A, 5-HT1B and 5-HT1D receptors. Naunyn Schmiedebergs Arch. Pharmacol. 1999, 359, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Solinas, M.; Tanda, G.; Wertheim, C.E.; Goldberg, S.R. Dopaminergic augmentation of delta-9-tetrahydrocannabinol (THC) discrimination: Possible involvement of D(2)-induced formation of anandamide. Psychopharmacology 2010, 209, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Romero, A.G.; Darlington, W.H.; McMillan, M.W. Synthesis of the Selective D2 Receptor Agonist PNU-95666E from d-Phenylalanine Using a Sequential Oxidative Cyclization Strategy. J. Org. Chem. 1997, 62, 6582–6587. [Google Scholar] [CrossRef]
- McCall, R.B.; Lookingland, K.J.; Bédard, P.J.; Huff, R.M. Sumanirole, a highly dopamine D2-selective receptor agonist: In vitro and in vivo pharmacological characterization and efficacy in animal models of Parkinson’s disease. J. Pharmacol. Exp. Ther. 2005, 314, 1248–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heier, R.F.; Dolak, L.A.; Duncan, J.N.; Hyslop, D.K.; Lipton, M.F.; Martin, I.J.; Mauragis, M.A.; Piercey, M.F.; Nichols, N.F.; Schreur, P.J.; et al. Synthesis and biological activities of (R)-5,6-dihydro-N,N-dimethyl-4H-imidazo[4,5,1-ij]quinolin-5-amine and its metabolites. J. Med. Chem. 1997, 40, 639–646. [Google Scholar] [CrossRef]
- Zou, M.F.; Keck, T.M.; Kumar, V.; Donthamsetti, P.; Michino, M.; Burzynski, C.; Schweppe, C.; Bonifazi, A.; Free, R.B.; Sibley, D.R.; et al. Novel Analogues of (R)-5-(Methylamino)-5,6-dihydro-4H-imidazo[4,5,1-ij]quinolin-2(1H)-one (Sumanirole) Provide Clues to Dopamine D2/D3 Receptor Agonist Selectivity. J. Med. Chem. 2016, 59, 2973–2988. [Google Scholar] [CrossRef]
- Villalón, C.M.; Ramírez-San Juan, E.; Sánchez-López, A.; Bravo, G.; Willems, E.W.; Saxena, P.R.; Centurión, D. Pharmacological profile of the vascular responses to dopamine in the canine external carotid circulation. Pharmacol. Toxicol. 2003, 92, 165–172. [Google Scholar] [CrossRef]
- Wilcox, R.E.; Huang, W.-H.; Brusniak, M.-Y.K.; Wilcox, D.M.; Pearlman, R.S.; Teeter, M.M.; DuRand, C.J.; Wiens, B.L.; Neve, K.A. CoMFA-Based Prediction of Agonist Affinities at Recombinant Wild Type versus Serine to Alanine Point Mutated D2 Dopamine Receptors. J. Med. Chem. 2000, 43, 3005–3019. [Google Scholar] [CrossRef]
- Martin, S.W.; Broadley, K.J. Renal vasodilatation by dopexamine and fenoldopam due to α1-adrenoceptor blockade. Br. J. Pharmacol. 1995, 115, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Ohlstein, E.H.; Zabko-Potapovich, B.; Berkowitz, B.A. The DA1 receptor agonist fenoldopam (SK & F 82526) is also an α2-adrenoceptor antagonist. Eur. J. Pharmacol. 1985, 118, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Nichols, A.J.; Ruffolo, R.R., Jr.; Brooks, D.P. The pharmacology of fenoldopam. Am. J. Hypertens. 1990, 3, 116S–119S. [Google Scholar] [CrossRef] [PubMed]
- Schetz, J.A.; Benjamin, P.S.; Sibley, D.R. Nonconserved residues in the second transmembrane-spanning domain of the D(4) dopamine receptor are molecular determinants of D(4)-selective pharmacology. Mol. Pharmacol. 2000, 57, 144–152. [Google Scholar]
- Shapiro, D.A.; Renock, S.; Arrington, E.; Chiodo, L.A.; Liu, L.X.; Sibley, D.R.; Roth, B.L.; Mailman, R. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003, 28, 1400–1411. [Google Scholar] [CrossRef] [Green Version]
- Lawler, C.P.; Prioleau, C.; Lewis, M.M.; Mak, C.; Jiang, D.; Schetz, J.A.; Gonzalez, A.M.; Sibley, D.R.; Mailman, R.B. Interactions of the novel antipsychotic aripiprazole (OPC-14597) with dopamine and serotonin receptor subtypes. Neuropsychopharmacology 1999, 20, 612–627. [Google Scholar] [CrossRef]
- Kroeze, W.K.; Hufeisen, S.J.; Popadak, B.A.; Renock, S.M.; Steinberg, S.; Ernsberger, P.; Jayathilake, K.; Meltzer, H.Y.; Roth, B.L. H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology 2003, 28, 519–526. [Google Scholar] [CrossRef]
- Shen, Y.; Monsma, F.J., Jr.; Metcalf, M.A.; Jose, P.A.; Hamblin, M.W.; Sibley, D.R. Molecular cloning and expression of a 5-hydroxytryptamine7 serotonin receptor subtype. J. Biol. Chem. 1993, 268, 18200–18204. [Google Scholar] [CrossRef]
- Kohen, R.; Metcalf, M.A.; Khan, N.; Druck, T.; Huebner, K.; Lachowicz, J.E.; Meltzer, H.Y.; Sibley, D.R.; Roth, B.L.; Hamblin, M.W. Cloning, characterization, and chromosomal localization of a human 5-HT6 serotonin receptor. J. Neurochem. 1996, 66, 47–56. [Google Scholar] [CrossRef]
- Boess, F.G.; Monsma, F.J., Jr.; Sleight, A.J. Identification of residues in transmembrane regions III and VI that contribute to the ligand binding site of the serotonin 5-HT6 receptor. J. Neurochem. 1998, 71, 2169–2177. [Google Scholar] [CrossRef]
- Gregori-Puigjané, E.; Setola, V.; Hert, J.; Crews, B.A.; Irwin, J.J.; Lounkine, E.; Marnett, L.; Roth, B.L.; Shoichet, B.K. Identifying mechanism-of-action targets for drugs and probes. Proc. Natl. Acad. Sci. USA 2012, 109, 11178–11183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, A.M.; Charifson, P.S.; Owens, C.E.; Kula, N.S.; McPhail, A.T.; Baldessarini, R.J.; Booth, R.G.; Wyrick, S.D. Conformational Analysis, Pharmacophore Identification, and Comparative Molecular Field Analysis of Ligands for the Neuromodulatory ς 3 Receptor. J. Med. Chem. 1994, 37, 4109–4117. [Google Scholar] [CrossRef] [PubMed]
- Freedman, S.B.; Patel, S.; Marwood, R.; Emms, F.; Seabrook, G.R.; Knowles, M.R.; McAllister, G. Expression and pharmacological characterization of the human D3 dopamine receptor. J. Pharmacol. Exp. Ther. 1994, 268, 417–426. [Google Scholar]
- Grundt, P.; Husband, S.L.; Luedtke, R.R.; Taylor, M.; Newman, A.H. Analogues of the dopamine D2 receptor antagonist L741, 626: Binding, function, and SAR. Bioorg. Med. Chem. Lett. 2007, 17, 745–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vangveravong, S.; Taylor, M.; Xu, J.; Cui, J.; Calvin, W.; Babic, S.; Luedtke, R.R.; Mach, R.H. Synthesis and characterization of selective dopamine D2 receptor antagonists. 2. Azaindole, benzofuran, and benzothiophene analogs of L-741, 626. Bioorg. Med. Chem. 2010, 18, 5291–5300. [Google Scholar] [CrossRef] [Green Version]
- Hirokawa, Y.; Morie, T.; Yamazaki, H.; Yoshida, N.; Kato, S. A novel series of N-(hexahydro-1,4-diazepin-6-yl) and N-(hexahydroazepin-3-yl)benzamides with high affinity for 5-HT3 and dopamine D2 receptors. Bioorg. Med. Chem. Lett. 1998, 8, 619–624. [Google Scholar] [CrossRef]
- Sautel, F.; Griffon, N.; Sokoloff, P.; Schwartz, J.C.; Launay, C.; Simon, P.; Costentin, J.; Schoenfelder, A.; Garrido, F.; Mann, A.; et al. Nafadotride, a potent preferential dopamine D3 receptor antagonist, activates locomotion in rodents. J. Pharmacol. Exp. Ther. 1995, 275, 1239–1246. [Google Scholar]
- Newman-Tancredi, A.; Gavaudan, S.; Conte, C.; Chaput, C.; Touzard, M.; Verrièle, L.; Audinot, V.; Millan, M.J. Agonist and antagonist actions of antipsychotic agents at 5-HT1A receptors: A [35S]GTPgammaS binding study. Eur. J. Pharmacol. 1998, 355, 245–256. [Google Scholar] [CrossRef]
- Grundt, P.; Prevatt, K.M.; Cao, J.; Taylor, M.; Floresca, C.Z.; Choi, J.K.; Jenkins, B.G.; Luedtke, R.R.; Newman, A.H. Heterocyclic analogues of N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)arylcarboxamides with functionalized linking chains as novel dopamine D3 receptor ligands: Potential substance abuse therapeutic agents. J. Med. Chem. 2007, 50, 4135–4146. [Google Scholar] [CrossRef]
- Newman, A.H.; Grundt, P.; Nader, M.A. Dopamine D3 receptor partial agonists and antagonists as potential drug abuse therapeutic agents. J. Med. Chem. 2005, 48, 3663–3679. [Google Scholar] [CrossRef]
- Keck, T.M.; John, W.S.; Czoty, P.W.; Nader, M.A.; Newman, A.H. Identifying Medication Targets for Psychostimulant Addiction: Unraveling the Dopamine D3 Receptor Hypothesis. J. Med. Chem. 2015, 58, 5361–5380. [Google Scholar] [CrossRef]
- Tang, L.; Todd, R.D.; Heller, A.; O´Malley, K.L. Pharmacological and functional characterization of D2, D3 and D4 dopamine receptors in fibroblast and dopaminergic cell lines. J. Pharmacol. Exp. Ther. 1994, 268, 495–502. [Google Scholar] [PubMed]
- MacKenzie, R.G.; VanLeeuwen, D.; Pugsley, T.A.; Shih, Y.H.; Demattos, S.; Tang, L.; Todd, R.D.; O´Malley, K.L. Characterization of the human dopamine D3 receptor expressed in transfected cell lines. Eur. J. Pharmacol. 1994, 266, 79–85. [Google Scholar] [CrossRef]
- Sleight, A.J.; Stam, N.J.; Mutel, V.; Vanderheyden, P.M. Radiolabelling of the human 5-HT2A receptor with an agonist, a partial agonist and an antagonist: Effects on apparent agonist affinities. Biochem. Pharmacol. 1996, 51, 71–76. [Google Scholar] [CrossRef]
- Maroteaux, L.; Saudou, F.; Amlaiky, N.; Boschert, U.; Plassat, J.L.; Hen, R. Mouse 5HT1B serotonin receptor: Cloning, functional expression, and localization in motor control centers. Proc. Natl. Acad. Sci. USA 1992, 89, 3020–3024. [Google Scholar] [CrossRef] [Green Version]
- Yoshio, R.; Taniguchi, T.; Itoh, H.; Muramatsu, I. Affinity of serotonin receptor antagonists and agonists to recombinant and native alpha1-adrenoceptor subtypes. Jpn. J. Pharmacol. 2001, 86, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Stark, D.; Piel, M.; Hübner, H.; Gmeiner, P.; Gründer, G.; Rösch, F. In vitro affinities of various halogenated benzamide derivatives as potential radioligands for non-invasive quantification of D(2)-like dopamine receptors. Bioorg. Med. Chem. 2007, 15, 6819–6829. [Google Scholar] [CrossRef]
- Dörfler, M.; Tschammer, N.; Hamperl, K.; Hübner, H.; Gmeiner, P. Novel D3 selective dopaminergics incorporating enyne units as nonaromatic catechol bioisosteres: Synthesis, bioactivity, and mutagenesis studies. J. Med. Chem. 2008, 51, 6829–6838. [Google Scholar] [CrossRef]
- Ricci, A.; Veglio, F.; Amenta, F. Radioligand binding characterization of putative dopamine D3 receptor in human peripheral blood lymphocytes with [3H]7-OH-DPAT. J. Neuroimmunol. 1995, 58, 139–144. [Google Scholar] [CrossRef]
- Brown, D.A.; Mishra, M.; Zhang, S.; Biswas, S.; Parrington, I.; Antonio, T.; Reith, M.E.; Dutta, A.K. Investigation of various N-heterocyclic substituted piperazine versions of 5/7-{[2-(4-aryl-piperazin-1-yl)-ethyl]-propyl-amino}-5,6,7,8-tetrahydro-naphthalen-2-ol: Effect on affinity and selectivity for dopamine D3 receptor. Bioorg. Med. Chem. 2009, 17, 3923–3933. [Google Scholar] [CrossRef] [Green Version]
- Maggio, R.; Scarselli, M.; Novi, F.; Corsini, G.U. Heterodimerization of G-Protein-Coupled Receptors Reveals an Unexpected Level of Pharmacological Diversity. Med. Chem. Res. 2004, 13, 25–33. [Google Scholar] [CrossRef]
- Stjernlöf, P.; Lin, C.-H.; Sonesson, C.; Svensson, K.; Smith, M.W. (Dipropylamino)-tetrahydronaphthofurans: Centrally acting serotonin agonists and dopamine agonists-antagonists. Bioorg. Med. Chem. Lett. 1997, 7, 2759–2764. [Google Scholar] [CrossRef]
- Chumpradit, S.; Kung, M.P.; Vessotskie, J.; Foulon, C.; Mu, M.; Kung, H.F. Iodinated 2-aminotetralins and 3-amino-1-benzopyrans: Ligands for dopamine D2 and D3 receptors. J. Med. Chem. 1994, 37, 4245–4250. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D.; Keating, G.M. Blonanserin: A review of its use in the management of schizophrenia. CNS Drugs 2010, 24, 65–84. [Google Scholar] [CrossRef] [PubMed]
- Ochi, T.; Sakamoto, M.; Minamida, A.; Suzuki, K.; Ueda, T.; Une, T.; Toda, H.; Matsumoto, K.; Terauchi, Y. Syntheses and properties of the major hydroxy metabolites in humans of blonanserin AD-5423, a novel antipsychotic agent. Bioorg. Med. Chem. Lett. 2005, 15, 1055–1059. [Google Scholar] [CrossRef]
- Hida, H.; Mouri, A.; Mori, K.; Matsumoto, Y.; Seki, T.; Taniguchi, M.; Yamada, K.; Iwamoto, K.; Ozaki, N.; Nabeshima, T.; et al. Blonanserin ameliorates phencyclidine-induced visual-recognition memory deficits: The complex mechanism of blonanserin action involving D3-5-HT2A and D1-NMDA receptors in the mPFC. Neuropsychopharmacology 2015, 40, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Tenjin, T.; Miyamoto, S.; Ninomiya, Y.; Kitajima, R.; Ogino, S.; Miyake, N.; Yamaguchi, N. Profile of blonanserin for the treatment of schizophrenia. Neuropsychiatr. Dis. Treat. 2013, 9, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Roth, B.L.; Craigo, S.C.; Choudhary, M.S.; Uluer, A.; Monsma, F.J., Jr.; Shen, Y.; Meltzer, H.Y.; Sibley, D.R. Binding of typical and atypical antipsychotic agents to 5-hydroxytryptamine-6 and 5-hydroxytryptamine-7 receptors. J. Pharmacol. Exp. Ther. 1994, 268, 1403–1410. [Google Scholar] [PubMed]
- Arnt, J.; Skarsfeldt, T. Do novel antipsychotics have similar pharmacological characteristics? A review of the evidence. Neuropsychopharmacology 1998, 18, 63–101. [Google Scholar] [CrossRef]
- Egan, C.T.; Herrick-Davis, K.; Teitler, M. Creation of a constitutively activated state of the 5-hydroxytryptamine2A receptor by site-directed mutagenesis: Inverse agonist activity of antipsychotic drugs. J. Pharmacol. Exp. Ther. 1998, 286, 85–90. [Google Scholar]
- Thomas, D.R.; Gittins, S.A.; Collin, L.L.; Middlemiss, D.N.; Riley, G.; Hagan, J.; Gloger, I.; Ellis, C.E.; Forbes, I.T.; Brown, A.M. Functional characterisation of the human cloned 5-HT7 receptor (long form); antagonist profile of SB-258719. Br. J. Pharmacol. 1998, 124, 1300–1306. [Google Scholar] [CrossRef] [Green Version]
- Fernández, J.; Alonso, J.M.; Andrés, J.I.; Cid, J.M.; Díaz, A.; Iturrino, L.; Gil, P.; Megens, A.; Sipido, V.K.; Trabanco, A.A. Discovery of new tetracyclic tetrahydrofuran derivatives as potential broad-spectrum psychotropic agents. J. Med. Chem. 2005, 48, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Schotte, A.; Janssen, P.F.; Gommeren, W.; Luyten, W.H.; Van Gompel, P.; Lesage, A.S.; De Loore, K.; Leysen, J.E. Risperidone compared with new and reference antipsychotic drugs: In vitro and in vivo receptor binding. Psychopharmacology 1996, 124, 57–73. [Google Scholar] [CrossRef]
- Lange, J.H.; Reinders, J.H.; Tolboom, J.T.; Glennon, J.C.; Coolen, H.K.; Kruse, C.G. Principal component analysis differentiates the receptor binding profiles of three antipsychotic drug candidates from current antipsychotic drugs. J. Med. Chem. 2007, 50, 5103–5108. [Google Scholar] [CrossRef] [PubMed]
- Rowley, M.; Bristow, L.J.; Hutson, P.H. Current and novel approaches to the drug treatment of schizophrenia. J. Med. Chem. 2001, 44, 477–501. [Google Scholar] [CrossRef]
- Millan, M.J.; Peglion, J.L.; Vian, J.; Rivet, J.M.; Brocco, M.; Gobert, A.; Newman-Tancredi, A.; Dacquet, C.; Bervoets, K.; Girardon, S.; et al. Functional correlates of dopamine D3 receptor activation in the rat in vivo and their modulation by the selective antagonist, (+)-S 14297: 1. Activation of postsynaptic D3 receptors mediates hypothermia, whereas blockade of D2 receptors elicits prolactin secretion and catalepsy. J. Pharmacol. Exp. Ther. 1995, 275, 885–898. [Google Scholar]
- Purohit, A.; Smith, C.; Herrick-Davis, K.; Teitler, M. Stable expression of constitutively activated mutant h5HT6 and h5HT7 serotonin receptors: Inverse agonist activity of antipsychotic drugs. Psychopharmacology 2005, 179, 461–469. [Google Scholar] [CrossRef]
- Seeman, P.; Corbett, R.; Van Tol, H.H. Atypical neuroleptics have low affinity for dopamine D2 receptors or are selective for D4 receptors. Neuropsychopharmacology 1997, 16, 93–110; discussion 111–135. [Google Scholar] [CrossRef] [Green Version]
- Bandyopadhyaya, A.; Rajagopalan, D.R.; Rath, N.P.; Herrold, A.; Rajagopalan, R.; Napier, T.C.; Tedford, C.E.; Rajagopalan, P. The synthesis and receptor binding affinities of DDD-016, a novel, potential, atypical antipsychotic. MedChemComm 2012, 3, 580–583. [Google Scholar] [CrossRef]
- Ablordeppey, S.Y.; Altundas, R.; Bricker, B.; Zhu, X.Y.; Kumar, E.V.; Jackson, T.; Khan, A.; Roth, B.L. Identification of a butyrophenone analog as a potential atypical antipsychotic agent: 4-[4-(4-chlorophenyl)-1,4-diazepan-1-yl]-1-(4-fluorophenyl)butan-1-one. Bioorg. Med. Chem. 2008, 16, 7291–7301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolós, J.; Anglada, L.; Gubert, S.; Planas, J.M.; Agut, J.; Príncep, M.; De la Fuente, N.; Sacristán, A.; Ortiz, J.A. 7-[3-(1-piperidinyl)propoxy]chromenones as potential atypical antipsychotics. 2. Pharmacological profile of 7-[3-[4-(6-fluoro-1, 2-benzisoxazol-3-yl)-piperidin-1-yl]propoxy]-3-(hydroxymeth yl)chromen -4-one (abaperidone, FI-8602). J. Med. Chem. 1998, 41, 5402–5409. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Tsai, K.C.; Xia, L. Pharmacophore identification of alpha(1A)-adrenoceptor antagonists. Bioorg. Med. Chem. Lett. 2005, 15, 657–664. [Google Scholar] [CrossRef]
- Jørgensen, M.; Jørgensen, P.N.; Christoffersen, C.T.; Jensen, K.G.; Balle, T.; Bang-Andersen, B. Discovery of novel α1-adrenoceptor ligands based on the antipsychotic sertindole suitable for labeling as PET ligands. Bioorg. Med. Chem. 2013, 21, 196–204. [Google Scholar] [CrossRef]
- Kristensen, J.L.; Püschl, A.; Jensen, M.; Risgaard, R.; Christoffersen, C.T.; Bang-Andersen, B.; Balle, T. Exploring the neuroleptic substituent in octoclothepin: Potential ligands for positron emission tomography with subnanomolar affinity for α(1)-adrenoceptors. J. Med. Chem. 2010, 53, 7021–7034. [Google Scholar] [CrossRef]
- Kołaczkowski, M.; Marcinkowska, M.; Bucki, A.; Pawłowski, M.; Mitka, K.; Jaśkowska, J.; Kowalski, P.; Kazek, G.; Siwek, A.; Wasik, A.; et al. Novel arylsulfonamide derivatives with 5-HT6/5-HT7 receptor antagonism targeting behavioral and psychological symptoms of dementia. J. Med. Chem. 2014, 57, 4543–4557. [Google Scholar] [CrossRef] [PubMed]
- Krogsgaard-Larsen, N.; Jensen, A.A.; Kehler, J. Novel 7-phenylsulfanyl-1,2,3,4,10,10a-hexahydro-pyrazino[1,2-a]indoles as dual serotonin 5-HT2C and 5-HT6 receptor ligands. Bioorg. Med. Chem. Lett. 2010, 20, 5431–5433. [Google Scholar] [CrossRef]
- Balle, T.; Perregaard, J.; Ramirez, M.T.; Larsen, A.K.; Søby, K.K.; Liljefors, T.; Andersen, K. Synthesis and structure-affinity relationship investigations of 5-heteroaryl-substituted analogues of the antipsychotic sertindole. A new class of highly selective alpha(1) adrenoceptor antagonists. J. Med. Chem. 2003, 46, 265–283. [Google Scholar] [CrossRef]
- Seeman, P. Antipsychotic drugs, dopamine receptors, and schizophrenia. Clin. Neurosci. Res. 2001, 1, 53–60. [Google Scholar] [CrossRef]
- Burstein, E.S.; Ma, J.; Wong, S.; Gao, Y.; Pham, E.; Knapp, A.E.; Nash, N.R.; Olsson, R.; Davis, R.E.; Hacksell, U.; et al. Intrinsic efficacy of antipsychotics at human D2, D3, and D4 dopamine receptors: Identification of the clozapine metabolite N-desmethylclozapine as a D2/D3 partial agonist. J. Pharmacol. Exp. Ther. 2005, 315, 1278–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasudevan, S.R.; Moore, J.B.; Schymura, Y.; Churchill, G.C. Shape-based reprofiling of FDA-approved drugs for the H1 histamine receptor. J. Med. Chem. 2012, 55, 7054–7060. [Google Scholar] [CrossRef]
- Sun, B.; Feng, D.; Chu, M.L.-H.; Fish, I.; Lovera, S.; Sands, Z.A.; Kelm, S.; Valade, A.; Wood, M.; Ceska, T.; et al. Crystal structure of dopamine D1 receptor in complex with G protein and a non-catechol agonist. Nat. Commun. 2021, 12, 3305. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Taylor, M.; Griffin, S.A.; McInnis, T.; Sumien, N.; Mach, R.H.; Luedtke, R.R. Evaluation of Substituted N-Phenylpiperazine Analogs as D3 vs. D2 Dopamine Receptor Subtype Selective Ligands. Molecules 2021, 26, 3182. [Google Scholar] [CrossRef] [PubMed]
- Kalani, M.Y.S.; Vaidehi, N.; Hall, S.E.; Trabanino, R.J.; Freddolino, P.L.; Kalani, M.A.; Floriano, W.B.; Kam, V.W.T.; Goddard, W.A., 3rd. The predicted 3D structure of the human D2 dopamine receptor and the binding site and binding affinities for agonists and antagonists. Proc. Natl. Acad. Sci. USA 2004, 101, 3815–3820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cover, K.K.; Mathur, B.N. Axo-axonic synapses: Diversity in neural circuit function. J. Comp. Neurol. 2021, 529, 2391–2401. [Google Scholar] [CrossRef]
- Borroto-Escuela, D.O.; Ambrogini, P.; Narvaez, M.; Di Liberto, V.; Beggiato, S.; Ferraro, L.; Fores-Pons, R.; Alvarez-Contino, J.E.; Lopez-Salas, A.; Mudò, G.; et al. Serotonin Heteroreceptor Complexes and Their Integration of Signals in Neurons and Astroglia—Relevance for Mental Diseases. Cells 2021, 10, 1902. [Google Scholar] [CrossRef]
- Chagraoui, A.; Boulain, M.; Juvin, L.; Anouar, Y.; Barrière, G.; Deurwaerdère, P.D. L-DOPA in Parkinson’s Disease: Looking at the “False” Neurotransmitters and Their Meaning. Int. J. Mol. Sci. 2020, 21, 294. [Google Scholar] [CrossRef] [Green Version]
- Cachope, R.; Cheer, J.F. Local control of striatal dopamine release. Front. Behav. Neurosci. 2014, 8, 188. [Google Scholar] [CrossRef]
- Fuxe, K.; Borroto-Escuela, D.; Romero-Fernandez, W.; Zhang, W.-B.; Agnati, L. Volume transmission and its different forms in the central nervous system. Chin. J. Integr. Med. 2013, 19, 323–329. [Google Scholar] [CrossRef]
- Zoli, M.; Torri, C.; Ferrari, R.; Jansson, A.; Zini, I.; Fuxe, K.; Agnati, L.F. The emergence of the volume transmission concept. Brain Res. Rev. 1998, 26, 136–147. [Google Scholar] [CrossRef]
- Heinrich, J.N.; Butera, J.A.; Carrick, T.; Kramer, A.; Kowal, D.; Lock, T.; Marquis, K.L.; Pausch, M.H.; Popiolek, M.; Sun, S.-C.; et al. Pharmacological comparison of muscarinic ligands: Historical versus more recent muscarinic M1-preferring receptor agonists. Eur. J. Pharmacol. 2009, 605, 53–56. [Google Scholar] [CrossRef]
- Fujio, M.; Togo, Y.; Tomozane, H.; Kuroita, T.; Morio, Y.; Katayama, J.; Matsumoto, Y. N-[[1-(2-phenylethyl)pyrrolidin-2-yl]methyl]cyclohexanecarboxamides as selective 5-HT1A receptor agonists. Bioorg. Med. Chem. Lett. 2000, 10, 509–512. [Google Scholar] [CrossRef]
- Perez, M.; Jorand-Lebrun, C.; Pauwels, P.J.; Pallard, I.; Halazy, S. Dimers of 5HT1 ligands preferentially bind to 5HT1B/1D receptor subtypes. Bioorg. Med. Chem. Lett. 1998, 8, 1407–1412. [Google Scholar] [CrossRef]
- Haadsma-Svensson, S.R.; Svensson, K.; Duncan, N.; Smith, M.W.; Lin, C.H. C-9 and N-substituted analogs of cis-(3aR)-(−)-2,3,3a,4,5,9b-hexahydro-3-propyl-1H-benz[e]indole-9-carboxamide: 5-HT1A receptor agonists with various degrees of metabolic stability. J. Med. Chem. 1995, 38, 725–734. [Google Scholar] [CrossRef]
- Kalkman, H.O.; Subramanian, N.; Hoyer, D. Extended radioligand binding profile of iloperidone: A broad spectrum dopamine/serotonin/norepinephrine receptor antagonist for the management of psychotic disorders. Neuropsychopharmacology 2001, 25, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Kongsamut, S.; Roehr, J.E.; Cai, J.; Hartman, H.B.; Weissensee, P.; Kerman, L.L.; Tang, L.; Sandrasagra, A. Iloperidone binding to human and rat dopamine and 5-HT receptors. Eur. J. Pharmacol. 1996, 317, 417–423. [Google Scholar] [CrossRef]
- Strupczewski, J.T.; Bordeau, K.J.; Chiang, Y.; Glamkowski, E.J.; Conway, P.G.; Corbett, R.; Hartman, H.B.; Szewczak, M.R.; Wilmot, C.A.; Helsley, G.C. 3-[[(Aryloxy)alkyl]piperidinyl]-1,2-benzisoxazoles as D2/5-HT2 antagonists with potential atypical antipsychotic activity: Antipsychotic profile of iloperidone (HP 873). J. Med. Chem. 1995, 38, 1119–1131. [Google Scholar] [CrossRef]


{kind=link}
| D1 | D2 | D3 | D4 | D5 | |
|---|---|---|---|---|---|
| agonist | A77636 SKF-81297 SKF-83959 | MLS1547 2 Rotigotine 3 Ropinirole 4 Pramipexole 4 PD 128907 4 PD168,077 7 A412997 8 | Rotigotine 3 Ropinirole 4 Pramipexole 4 PD 128907 4 A412997 8 [3H]PD128907 9 | Rotigotine 3 PD168,077 7 A412997 8 | |
| antagonist | SKF-83566 1 SCH-23390 1 Ecopipam 1 [125I]SCH23982 1,9 | pipotiazine perospirone 5 raclopride 3 ML321 Prochlorperazine 4 Sulpiride 5 NGB 2904 6 | Perospirone 5 Raclopride 3 Prochlorperazine 4 Sulpiride 5 S33084 NGB 2904 6 SB 277011-A (+)-S-14297 | Perospirone 5 Sulpiride 5 sonepiprazole L745870 A-381393 L741742 ML398 [125I]L750667 9 [3H]NGD941 9 | SKF-83566 1 SCH-23390 1 Ecopipam 1 [125I]SCH23982 1,9 |
| CNS Area | DR Presence | α2-AR Presence | 5-HT Presence | |
|---|---|---|---|---|
| Cerebral cortex | D1-like D2-like | α2C-AR | 5-HT2 5-HT4 5-HT6 5-HT1A 5-HT1B 5 5-HT1E 5-HT1F 5-HT5A | |
| Amygdala | D1-like | α2C-AR α2A-AR2 | 5-HT2C 5-HT6 5-HT1B 5 | |
| Substantia nigra | pars compacta | D2 DR | α2C-AR | 5-HT4 5-HT1B 6 5-HT1D 6 5-HT1F 6 |
| pars reticularis | 5-HT4 5-HT1B 6 5-HT1D 6 5-HT1F 6 | |||
| Striatum (Caudate-putamen) | D1 DR D2 DR D3 DR | α2C-AR 1 | 5-HT2A/2C 5-HT4 1 5-HT6 5-HT1B 2 5-HT1D 1 5-HT1F 7 | |
| Globus pallidus | D2-like | α2C-AR 1 | 5-HT4 1 5-HT1B 5-HT1D 1 5-HT1F | |
| Ncl. accumbens | D1 DR | 5-HT2A/2C 5-HT6 5-HT1B 2 | ||
| Hippocampus (without further specification) | D5 DR D4 DR | α2C-AR α2A-AR 2 α2B-AR 2 | 5-HT4 5-HT6 5-HT7 5-HT1A 5-HT1F 5-HT5A 2 | |
| CA1 | D1-like D2-like | 5-HT4 5-HT1A 5-HT1B 2 5-HT1E 5-HT5A 2 | ||
| CA3 | D1-like D2-like | 5-HT1E 5-HT5A 2 | ||
| Thalamus | D1 DR | α2B-AR α2C-AR2 | 5-HT2A 5-HT6 5-HT7 | |
| Ncl. subthalamicus | D1 DR | α2C-AR 1 | 5-HT1B 2 | |
| Hypothalamus | D5 DR D3 DR | α2A-AR 2 | 5-HT2C 3 5-HT6 5-HT7 4 5-HT1A 5-HT1B 5 5-HT5 4 | |
| Olfactory tubercle | D3 DR | α2C-AR | 5-HT2A/2C 5-HT6 | |
| Midbrain | D4 DR | α2A-AR 2 α2C-AR 2 | ||
| Ventral tegmental area | D2 DR | |||
| Cerebellum | D3 DR D4 DR | α2A-AR 2 α2B-AR 2 | 5-HT6 5-HT1B 2 5-HT5A 2 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Myslivecek, J. Dopamine and Dopamine-Related Ligands Can Bind Not Only to Dopamine Receptors. Life 2022, 12, 606. https://doi.org/10.3390/life12050606
Myslivecek J. Dopamine and Dopamine-Related Ligands Can Bind Not Only to Dopamine Receptors. Life. 2022; 12(5):606. https://doi.org/10.3390/life12050606
Chicago/Turabian StyleMyslivecek, Jaromir. 2022. "Dopamine and Dopamine-Related Ligands Can Bind Not Only to Dopamine Receptors" Life 12, no. 5: 606. https://doi.org/10.3390/life12050606
APA StyleMyslivecek, J. (2022). Dopamine and Dopamine-Related Ligands Can Bind Not Only to Dopamine Receptors. Life, 12(5), 606. https://doi.org/10.3390/life12050606