
Dithymoquinone Analogues as Potential Candidate(s) for Neurological Manifestation Associated with COVID-19: A Therapeutic Strategy for Neuro-COVID

 , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
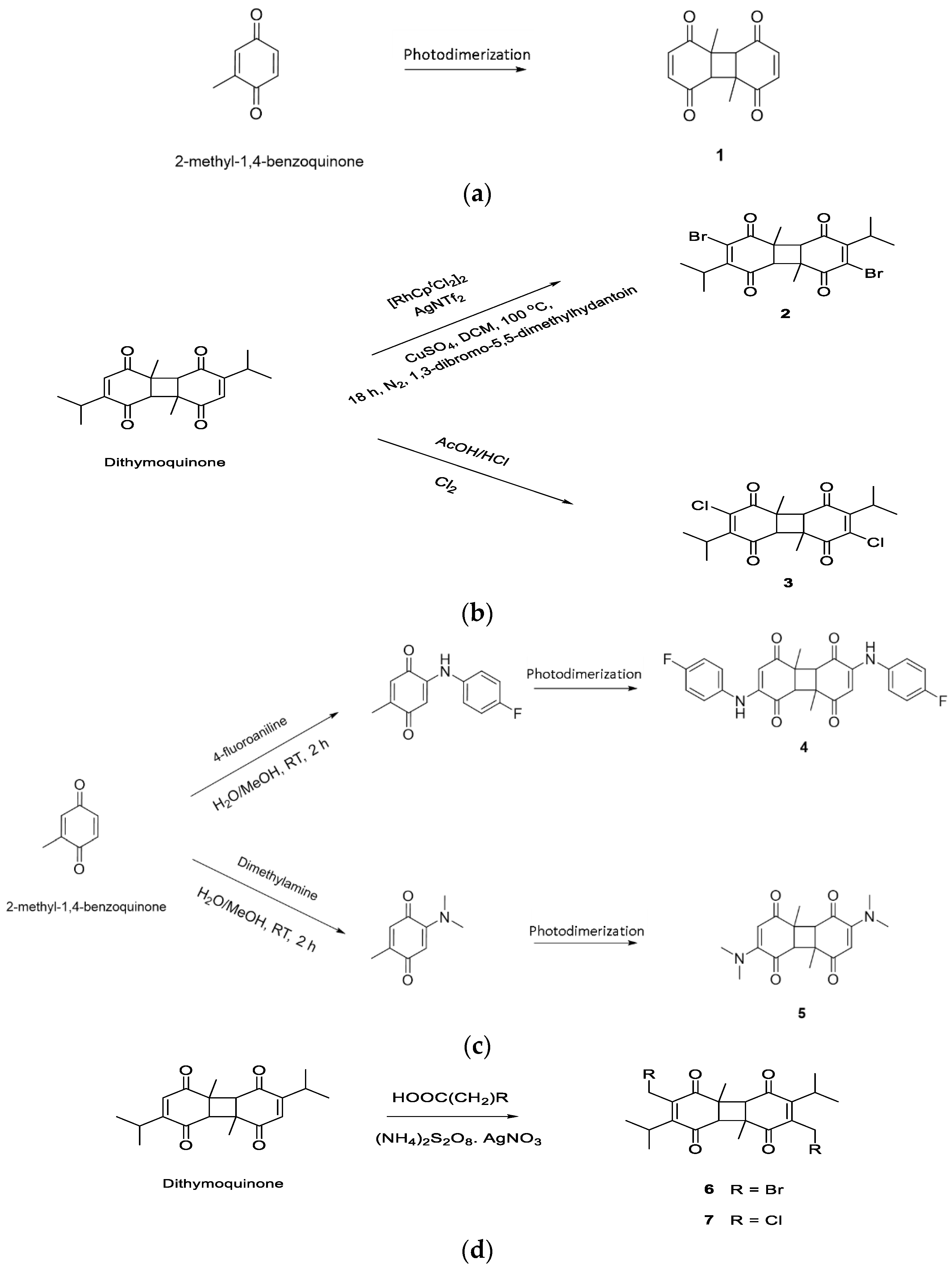
2.1. Designing DTQ Analogues
2.2. Physicochemical Parameters and Toxicity Prediction
2.3. Molecular Docking and Interaction Analysis
2.3.1. Target Protein Preparation
2.3.2. Ligand Preparation
2.3.3. Molecular Docking
2.4. Molecular Dynamics Simulation Analysis
3. Results and Discussion
3.1. DTQ Analogues Designing
3.2. Physicochemical Properties and Toxicity Potential Prediction of DTQ Analogues
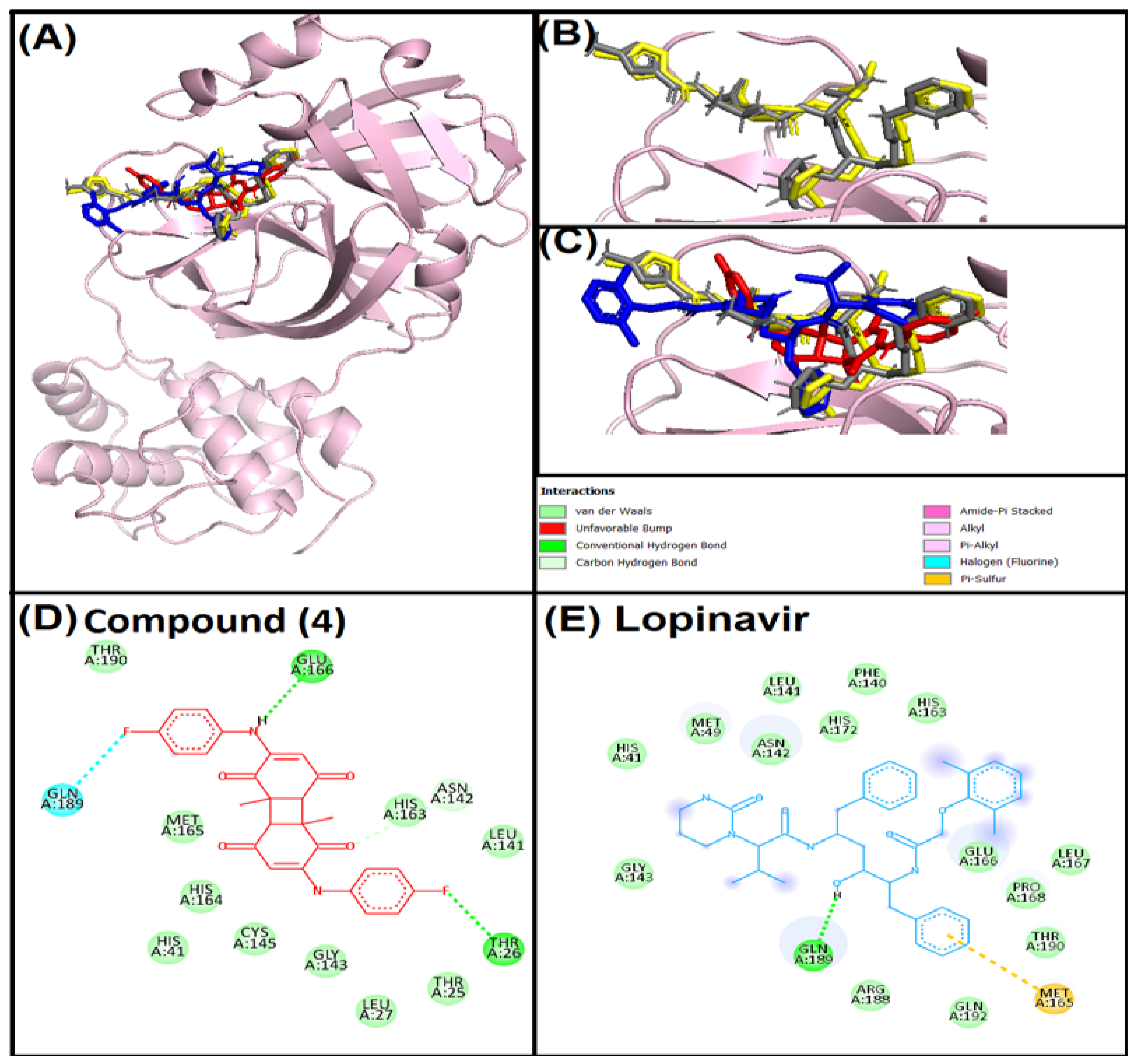
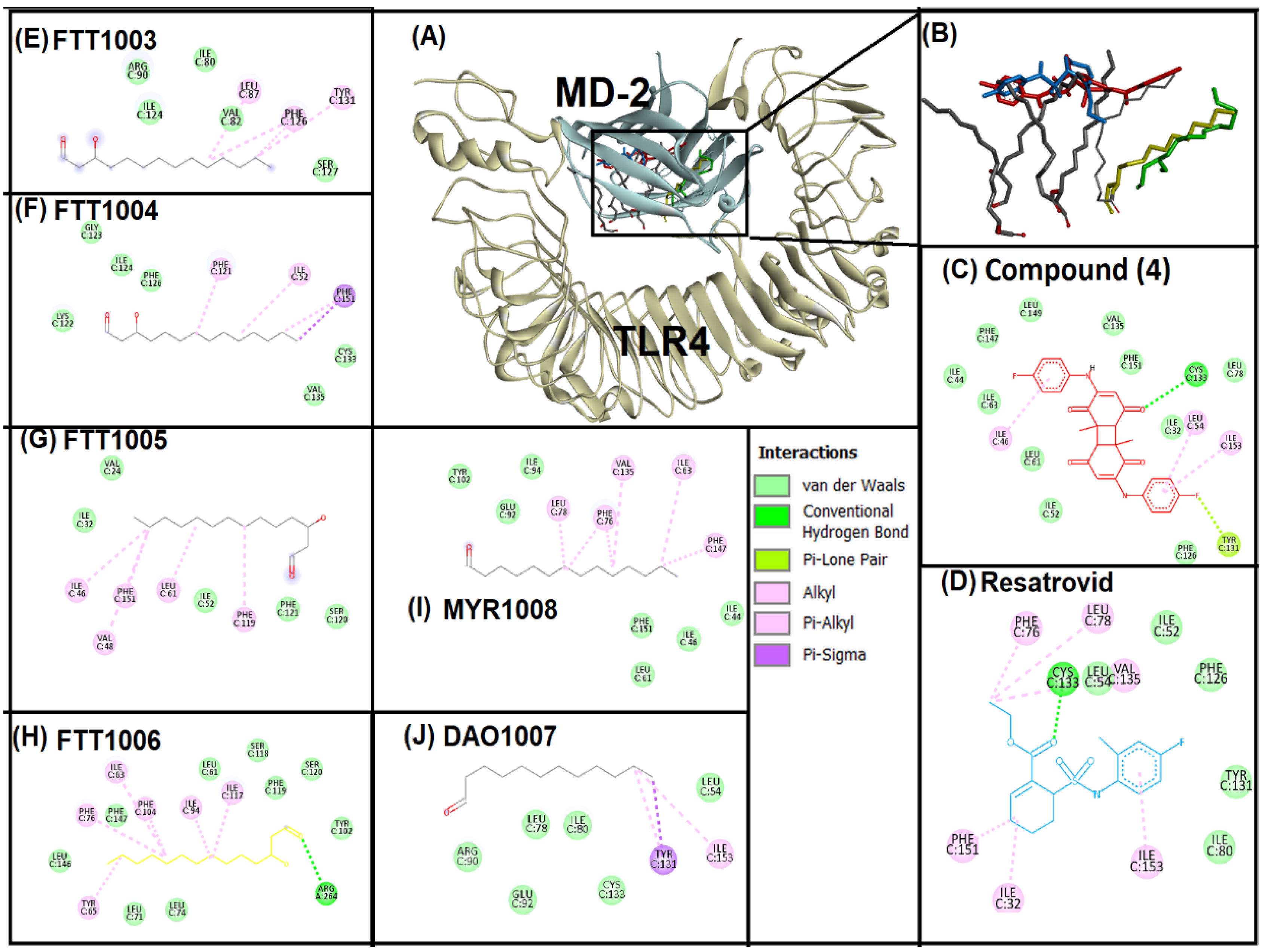
3.3. Molecular Docking Analysis
3.4. Analysis of Interaction of Compound (4) with Target Proteins
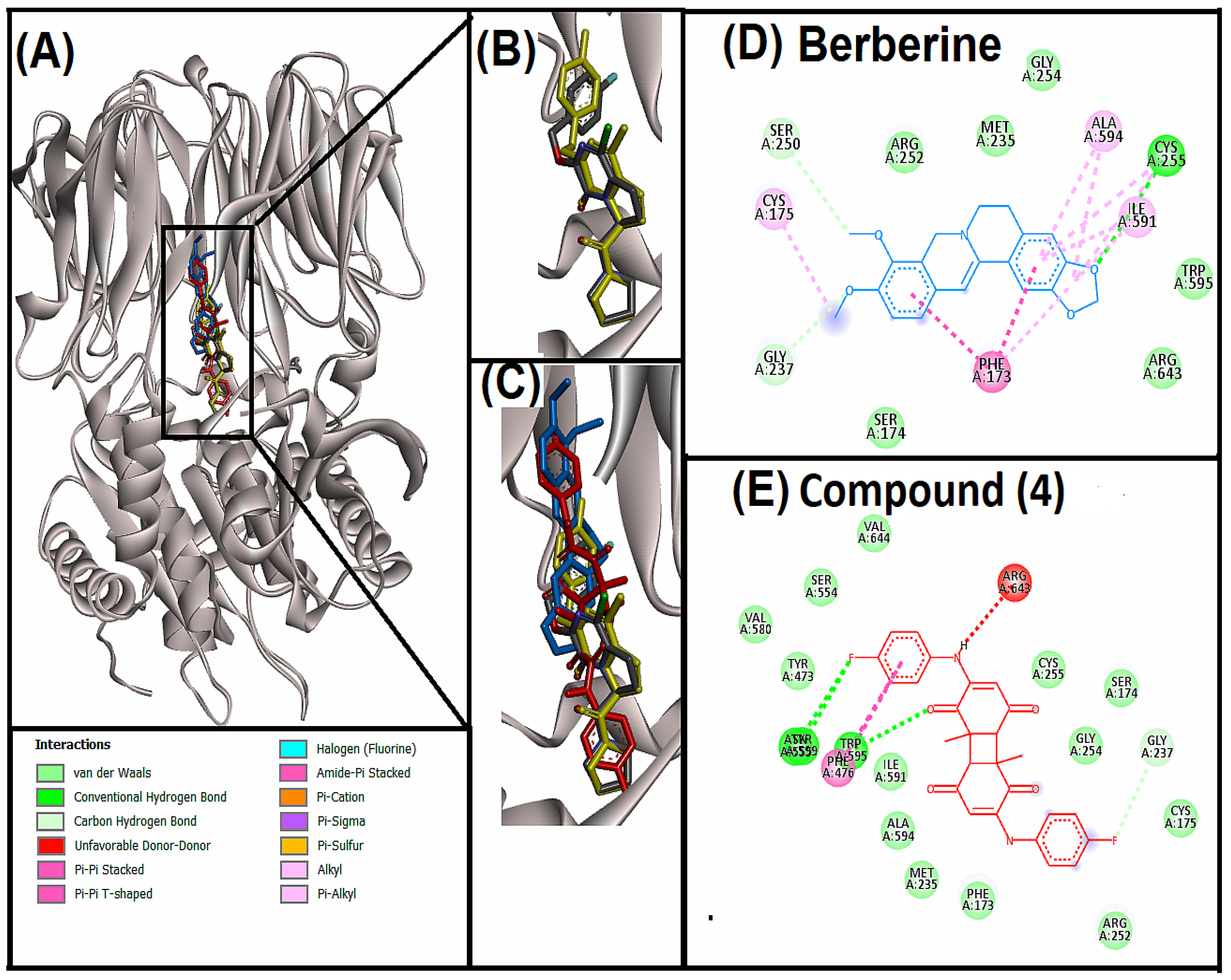
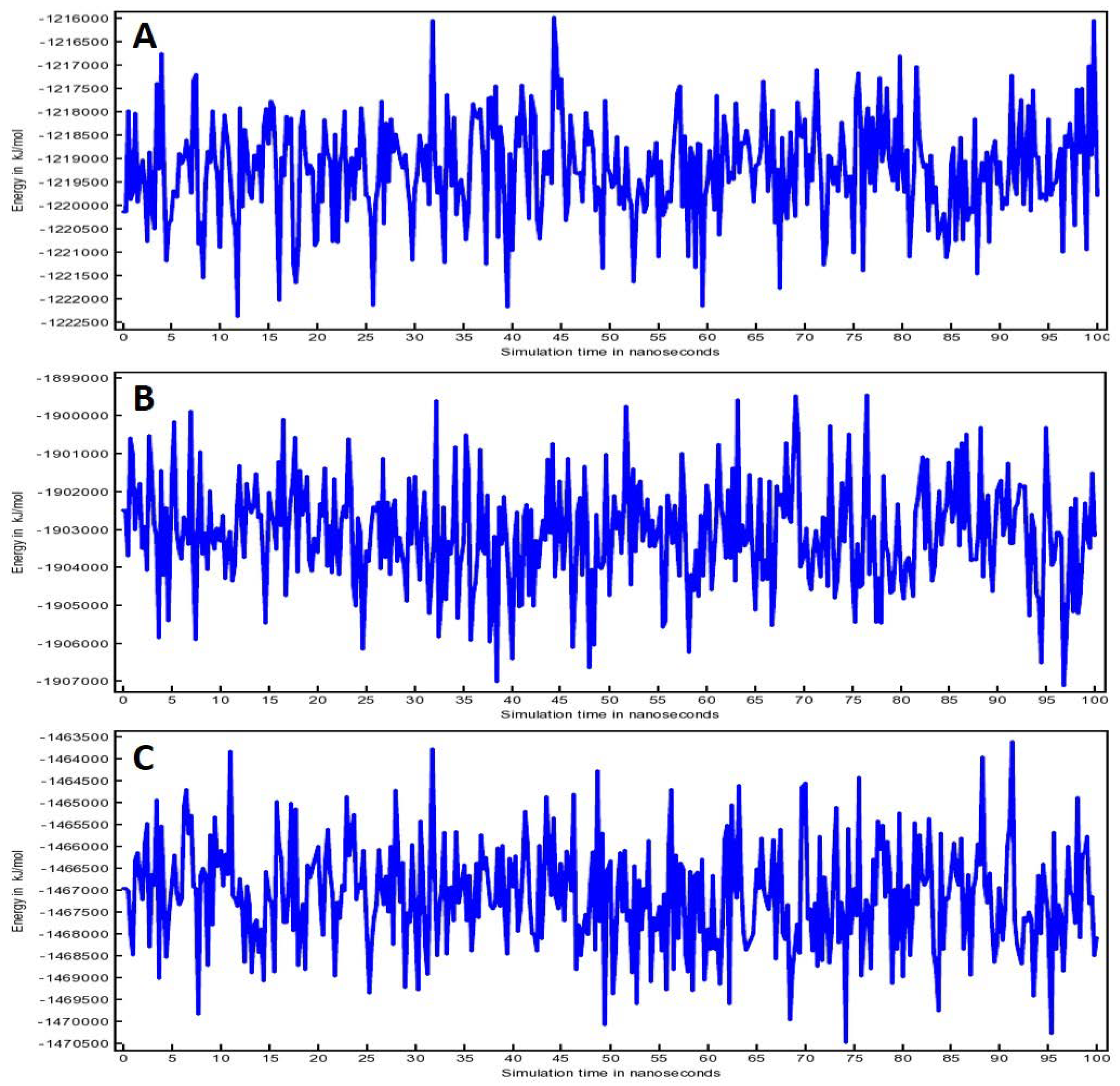
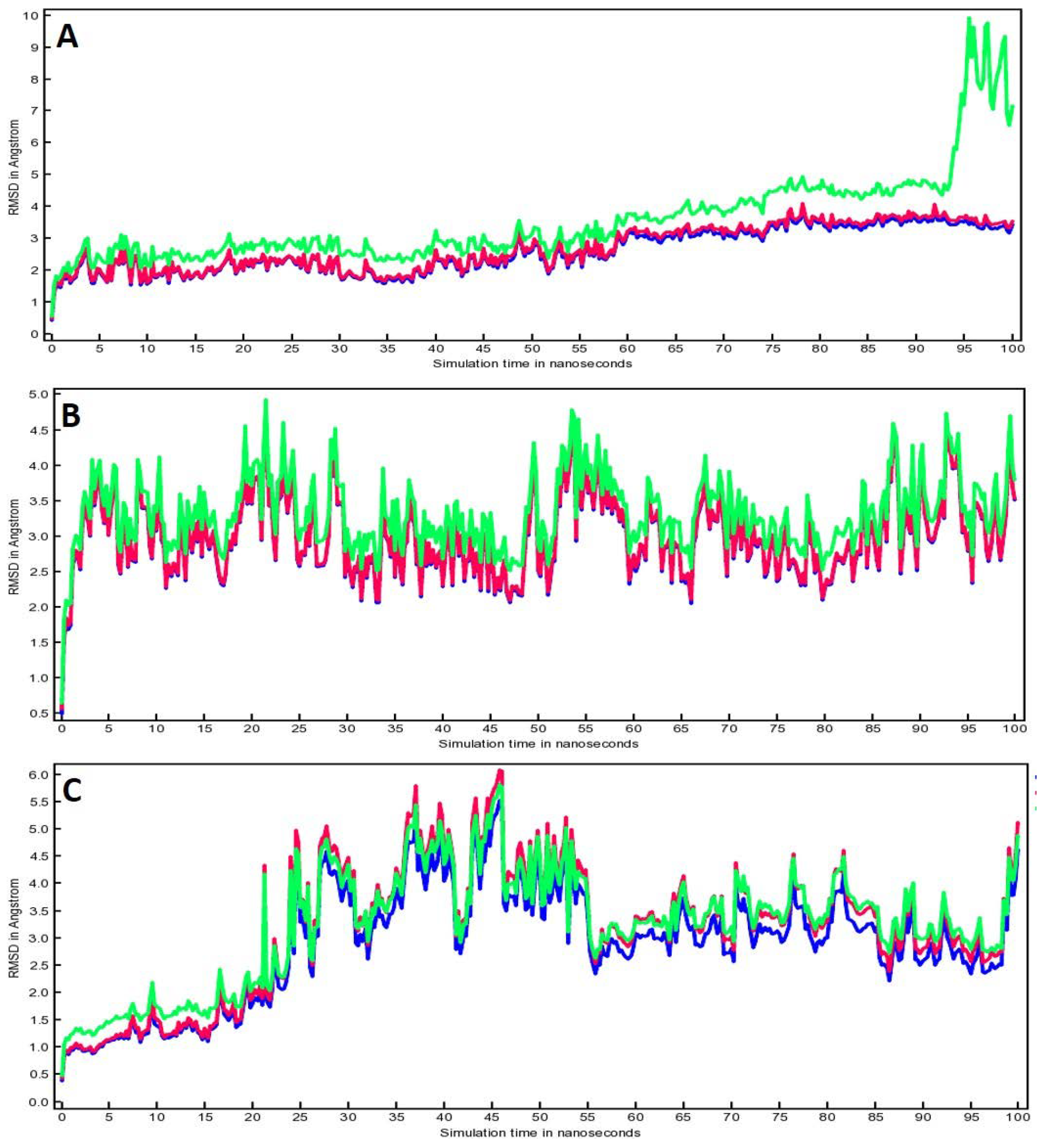
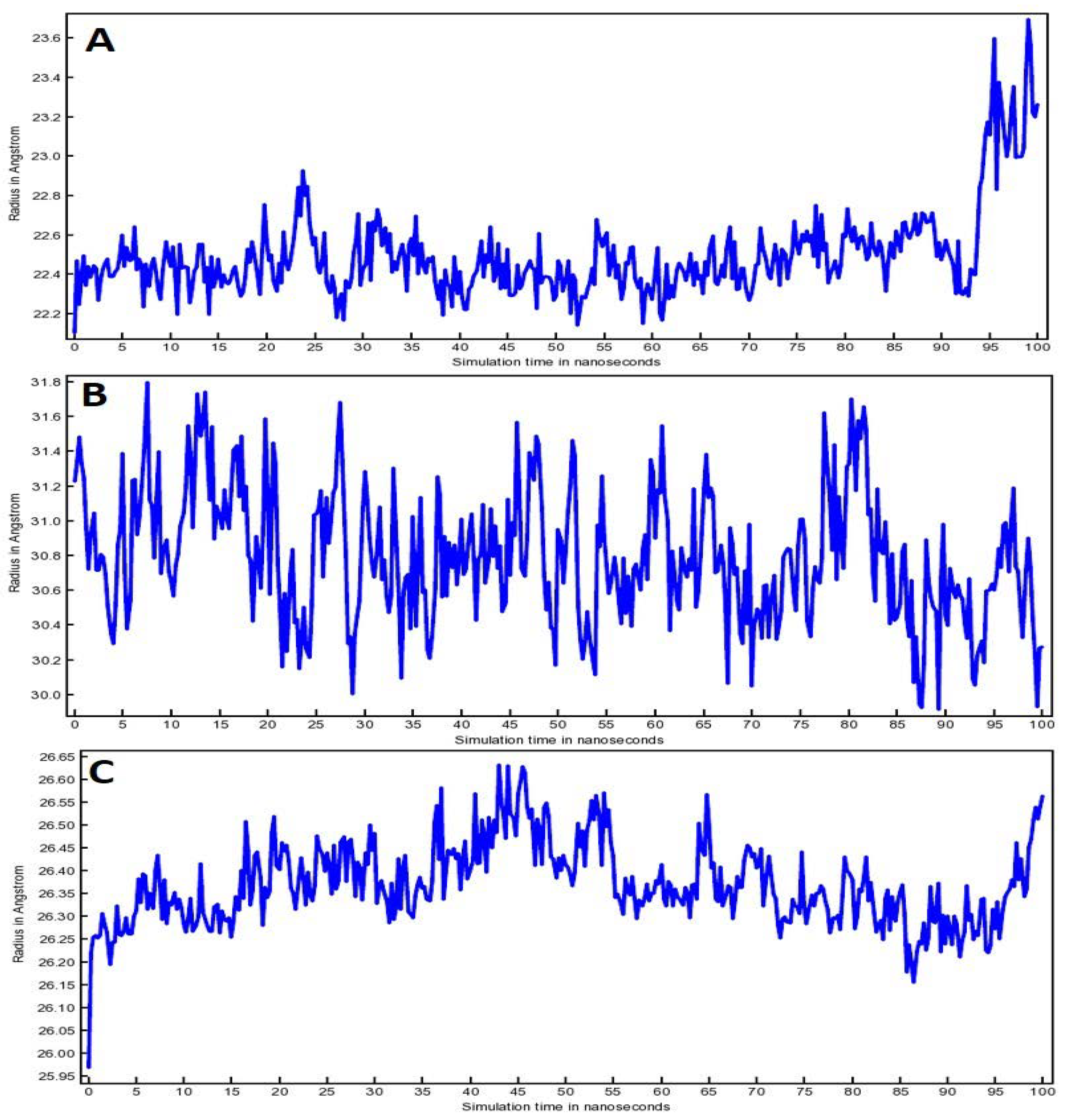
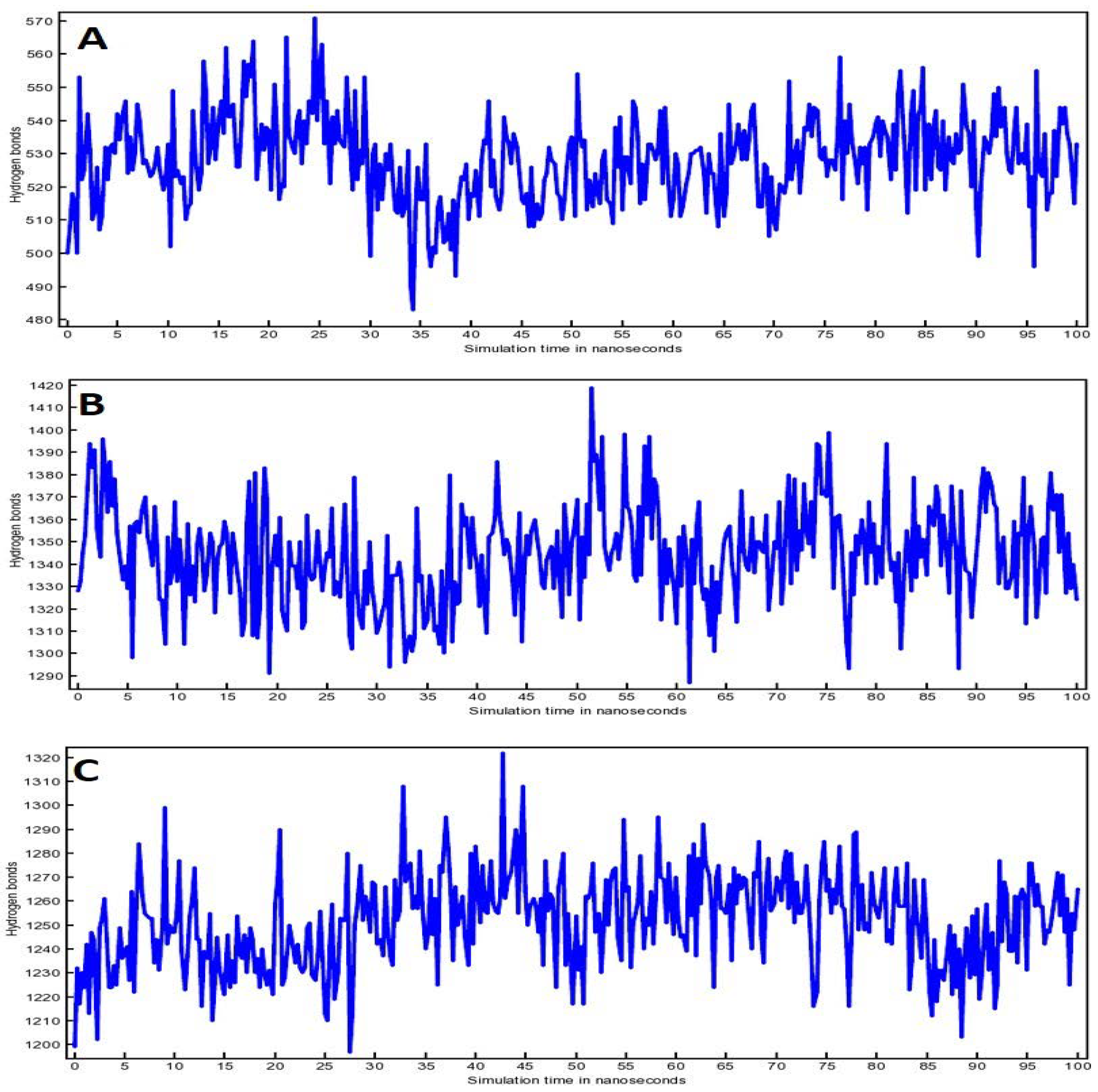
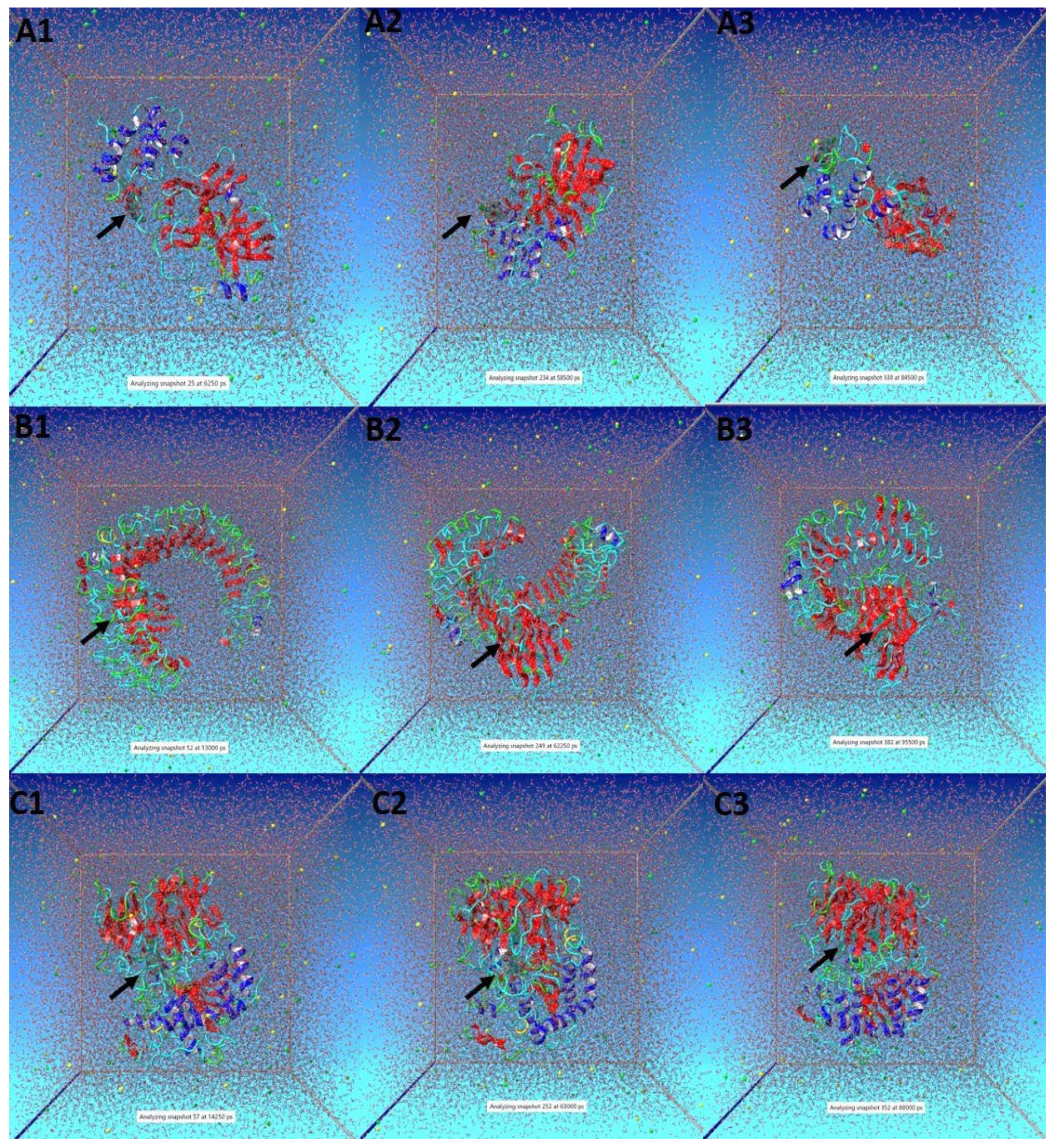
3.5. Molecular Dynamic (MD) Simulation Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Prasad, A.; Prasad, M. Single virus targeting multiple organs: What we know and where we are heading? Front. Med. 2020, 7, 370. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, X.; Chen, Z.; Duan, J.; Hashimoto, K.; Yang, L.; Liu, C.; Yang, C. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav. Immun. 2020, 87, 18–22. [Google Scholar] [CrossRef]
- Mao, L.; Wang, M.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; Li, Y.; Jin, H.; et al. Neurological manifestations of hospitalized patients with COVID-19 in Wuhan, China: A retrospective case series study. SSRN Electron. J. 2020, 77, 1–9. [Google Scholar] [CrossRef]
- Kaushik, D.; Bhandari, R.; Kuhad, A. TLR4 as a therapeutic target for respiratory and neurological complications of SARS-CoV-2. Expert Opin. Ther. Targets 2021, 25, 491–508. [Google Scholar] [CrossRef]
- Conte, C. Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4. Int. J. Mol. Sci. 2021, 22, 7135. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Sohn, K.M.; Lee, S.G.; Kim, H.J.; Cheon, S.; Jeong, H.; Lee, J.; Kim, I.S.; Silwal, P.; Kim, Y.J.; Paik, S.; et al. COVID-19 patients upregulate toll-like receptor 4-mediated inflammatory signaling that mimics bacterial sepsis. J. Korean Med. Sci. 2020, 35, e343. [Google Scholar] [CrossRef]
- Rizvi, S.M.D.; Hussain, T.; Moin, A.; Dixit, S.R.; Mandal, S.P.; Adnan, M.; Jamal, Q.M.S.; Sharma, D.C.; Alanazi, A.S.; Unissa, R. Identifying the Most Potent Dual-Targeting Compound(s) against 3CLprotease and NSP15exonuclease of SARS-CoV-2 from Nigella sativa: Virtual Screening via Physicochemical Properties, Docking and Dynamic Simulation Analysis. Processes 2021, 9, 1814. [Google Scholar] [CrossRef]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun. Biol. 2021, 4, 93. [Google Scholar] [CrossRef]
- Ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Yang, L.; Zhang, X.; Zhang, Q.; Yang, Z.; Liu, Y.; Wei, S.; Liu, W. Discovery of Potential Flavonoid Inhibitors against COVID-19 3CL Proteinase Based on Virtual Screening Strategy. Front. Mol. Biosci. 2020, 7, 556481. [Google Scholar] [CrossRef]
- Pérard-viret, J.; Quteishat, L.; Alsalim, R. Structural insights into coronavirus entry. Adv. Virus Res. 2020, 105, 94–95. [Google Scholar]
- Chen, R.; Wang, K.; Yu, J.; Howard, D.; French, L.; Chen, Z.; Wen, C.; Xu, Z. The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in the human and mouse brains. Front. Neurol. 2021, 11, 573095. [Google Scholar] [CrossRef]
- Svarcbahs, R.; Julku, U.H.; Norrbacka, S.; Myöhänen, T.T. Removal of prolyl oligopeptidase reduces alpha-synuclein toxicity in cells and in vivo. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Johnson-Ajinwo, O.R.; Ullah, I.; Mbye, H.; Richardson, A.; Horrocks, P.; Li, W.W. The synthesis and evaluation of thymoquinone analogues as anti-ovarian cancer and antimalarial agents. Bioorg. Med. Chem. Lett. 2018, 28, 1219–1222. [Google Scholar] [CrossRef]
- Myers, A.L.; Zhang, Y.P.; Kramer, M.A.; Bornmann, W.G.; Kaseb, A.; Yang, P.; Tran, H.T. A practical synthesis and X-ray crystallographic analysis of dithymoquinone, a photodimer of thymoquinone. Lett. Org. Chem. 2012, 9, 762. [Google Scholar] [CrossRef] [Green Version]
- Jardim, G.A.; Bower, J.F.; da Silva Junior, E.N. Rh-Catalyzed Reactions of 1, 4-Benzoquinones with Electrophiles: C–H Iodination, Bromination, and Phenylselenation. Org. Lett. 2016, 18, 4454–4457. [Google Scholar] [CrossRef] [Green Version]
- Ulfa, S.M.; Sholikhah, S.; Utomo, E.P. Synthesis of Thymoquinone derivatives and its activity analysis: In-silico approach. In AIP Conference Proceedings; AIP Publishing LLC: New York, NY, USA, 2017; Volume 1823, p. 020102. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Del. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Abraham, M.H.; Le, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B.; Cooper, I. Rate-Limited Steps of Human Oral Absorption and QSAR Studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef]
- Rizvi, S.M.D.; Shakil, S.; Haneef, M. A simple click by click protocol to perform docking: AutoDock 4.2 made easy for non-bioinformaticians. EXCLI J. 2013, 12, 831–857. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Dunbrack, R.L., Jr.; Hooft, R.W.; Krieger, B. Assignment of protonation states in proteins and ligands: Combining pKa prediction with hydrogen bonding network optimization. Methods Mol. Biol. 2012, 819, 405–421. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Kelder, J.; Grootenhuis, P.D.J.; Bayada, D.M.; Delbressine, L.P.C.; Ploemen, J.-P. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Van de Waterbeemd, H.; Camenish, G.; Folkers, G.; Chretien, J.R.; Raevsky, O.A. Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding characteristics. J. Drug Target. 1998, 6, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Abdusalam, A.A.A.; Murugaiyah, V. Identification of Potential Inhibitors of 3CL Protease of SARS-CoV-2 from ZINC Database by Molecular Docking-Based Virtual Screening. Front. Mol. Biosci. 2020, 7, 603037. [Google Scholar] [CrossRef]
- Choudhary, M.I.; Shaikh, M.; Tul-Wahab, A.; Ur-Rahman, A. In silico identification of potential inhibitors of key SARS-CoV-2 3CL hydrolase (Mpro) via molecular docking, MMGBSA predictive binding energy calculations, and molecular dynamics simulation. PLoS ONE 2020, 15, e0235030. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Yin, H.; Wang, X.; et al. SARS-CoV-2 spike protein interacts with and activates TLR41. Cell Res. 2021, 31, 818–820. [Google Scholar] [CrossRef]
- Espinoza-Culupú, A.; Vázquez-Ramírez, R.; Farfán-López, M.; Mendes, E.; Notomi Sato, M.; da Silva Junior, P.I.; Borges, M.M. Acylpolyamine Mygalin as a TLR4 Antagonist Based on Molecular Docking and In Vitro Analyses. Biomolecules 2020, 10, 1624. [Google Scholar] [CrossRef]
- Li, H.; Peng, Y.; Lin, C.; Zhang, X.; Zhang, T.; Wang, Y.; Li, Y.; Wu, S.; Wang, H.; Hutchinson, M.R.; et al. Nicotine and its metabolite cotinine target MD2 and inhibit TLR4 signaling. Innovation 2021, 2, 100111. [Google Scholar] [CrossRef] [PubMed]
- Savolainen, M.H.; Yan, X.; Myöhänen, T.T.; Huttunen, H.J. Prolyl oligopeptidase enhances α-synuclein dimerization via direct protein-protein interaction. J. Biol. Chem. 2015, 290, 5117–5126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babkova, K.; Korabecny, J.; Soukup, O.; Nepovimova, E.; Jun, D.; Kuca, K. Prolyl oligopeptidase and its role in the organism: Attention to the most promising and clinically relevant inhibitors. Future Med. Chem. 2017, 9, 1015–1038. [Google Scholar] [CrossRef]
- Silva-Aguiar, R.P.; Peruchetti, D.B.; Rocco, P.; Schmaier, A.H.; Silva, P.E.; Martins, M.A.; Carvalho, V.F.; Pinheiro, A.; Caruso-Neves, C. Role of the renin-angiotensin system in the development of severe COVID-19 in hypertensive patients. American journal of physiology. Lung Cellul. Mol. Physiol. 2020, 319, L596–L602. [Google Scholar] [CrossRef]
- Kumar, R.; Bavi, R.; Jo, M.G.; Arulalapperumal, V.; Baek, A.; Rampogu, S.; Kim, M.O.; Lee, K.W. New compounds identified through in silico approaches reduce the α-synuclein expression by inhibiting prolyl oligopeptidase In Vitro. Sci. Rep. 2017, 7, 10827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Abbasi, H.W.; Shahid, S.; Gul, S.; Abbasi, S.W. Molecular docking, simulation and MM-PBSA studies of nigella sativa compounds: A computational quest to identify potential natural antiviral for COVID-19 treatment. J. Biomol. Struct. Dyn. 2021, 39, 4225–4233. [Google Scholar] [CrossRef]
- Esharkawy, E.R.; Almalki, F.; Hadda, T.B. In Vitro potential antiviral SARS-CoV-19- activity of natural product thymohydroquinone and dithymoquinone from Nigella sativa. Bioorg. Chem. 2022, 120, 105587. [Google Scholar] [CrossRef]
- Elsharkawy, E.R.; Abdallah, E.M.; Abo Markb, A. Potential Cytotoxic, Antifungal, and Antioxidant Activity of Dithymoquinone and Thymoquinone. J. Hunan Univ. Nat. Sci. 2021, 48, 90–99. [Google Scholar]
- Crooks, P.A.; Worthen, D.R.; Ghosheh, O.A. Use of the Naturally-Occurring Quinones Thymoquinone and Dithymoquinone as Antineoplastic and Cytotoxic Agents. U.S. Patent 6,218,434, 17 April 2001. [Google Scholar]
- Xing, K.; Zhang, J.; Han, Y.; Tong, T.; Liu, D.; Zhao, L. Design, Synthesis and Bioactivity Evaluation of 4,6-Disubstituted Pyrido [3,2-d]pyrimidine Derivatives as Mnk and HDAC Inhibitors. Molecules 2020, 25, 4318. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DTQ Analogues /Control | Physicochemical Properties | Toxicity Potential | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % Abs | TPSA | M.W. | cLogP | H-acc. | H-don. | R.B. | L.V. | Mut. | Tum. | Reprod. | Irrit. | |
| Rule | <500 | ≤5 | <10 | <5 | ≤10 | ≤1 | ||||||
| Comp. (1) | 85.43 | 68.3 | 244.2 | 0.5 | 4.0 | 0.0 | 0.0 | 0 | None | None | None | None |
| Comp. (2) | 85.43 | 68.3 | 486.2 | 3.8 | 4.0 | 0.0 | 2.0 | 0 | None | None | None | None |
| Comp. (3) | 85.43 | 68.3 | 397.3 | 3.6 | 4.0 | 0.0 | 2.0 | 0 | High | None | None | None |
| Comp. (4) | 77.15 | 92.3 | 462.5 | 3.0 | 6.0 | 2.0 | 4.0 | 0 | None | None | None | None |
| Comp. (5) | 83.19 | 74.8 | 330.4 | −0.1 | 6.0 | 0.0 | 2.0 | 0 | None | None | None | None |
| Comp. (6) | 85.43 | 68.3 | 514.3 | 4.3 | 4.0 | 0.0 | 4.0 | 1 | High | High | High | None |
| Comp. (7) | 85.43 | 68.3 | 425.3 | 4.1 | 4.0 | 0.0 | 4.0 | 0 | Low | High | High | None |
| Comp. (8) | 85.43 | 68.3 | 328.4 | 2.7 | 4.0 | 0.0 | 2.0 | 0 | None | None | None | None |
| Lopinavir | 67.6 | 120.0 | 628.8 | 4.8 | 9.0 | 4.0 | 15.0 | 2 | None | None | None | High |
| Resatrovid | 81.12 | 80.8 | 361.8 | 2.8 | 5.0 | 1.0 | 5.0 | 0 | High | None | High | None |
| Berberine | 94.71 | 41.4 | 338.4 | 0.9 | 5.0 | 1.0 | 2.0 | 0 | Low | Low | None | None |
| DTQ Analogues /Control | 3CLpro | TLR-4 | PREP |
|---|---|---|---|
| Comp. (1) | −7.2 kcal/mol | −7.3 kcal/mol | −7.3 kcal/mol |
| Comp. (2) | −7.6 kcal/mol | −8.4 kcal/mol | −7.6 kcal/mol |
| Comp. (3) | −7.7 kcal/mol | −8.4 kcal/mol | −7.6 kcal/mol |
| Comp. (4) | −8.5 kcal/mol | −10.8 kcal/mol | −9.5 kcal/mol |
| Comp. (5) | −6.3 kcal/mol | −7 kcal/mol | −7 kcal/mol |
| Comp. (6) | −7.4 kcal/mol | −8.2 kcal/mol | −7.2 kcal/mol |
| Comp. (7) | −7.4 kcal/mol | −8.1 kcal/mol | −7.4 kcal/mol |
| Comp. (8) | −7.4 kcal/mol | −8.3 kcal/mol | −7.9 kcal/mol |
| Lopinavir | −8.4 kcal/mol | - | - |
| Resatrovid | - | −7.3 kcal/mol | - |
| Berberine | - | - | −7.5 kcal/mol |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moin, A.; Huwaimel, B.; Alobaida, A.; Break, M.K.B.; Iqbal, D.; Unissa, R.; Jamal, Q.M.S.; Hussain, T.; Sharma, D.C.; Rizvi, S.M.D. Dithymoquinone Analogues as Potential Candidate(s) for Neurological Manifestation Associated with COVID-19: A Therapeutic Strategy for Neuro-COVID. Life 2022, 12, 1076. https://doi.org/10.3390/life12071076
Moin A, Huwaimel B, Alobaida A, Break MKB, Iqbal D, Unissa R, Jamal QMS, Hussain T, Sharma DC, Rizvi SMD. Dithymoquinone Analogues as Potential Candidate(s) for Neurological Manifestation Associated with COVID-19: A Therapeutic Strategy for Neuro-COVID. Life. 2022; 12(7):1076. https://doi.org/10.3390/life12071076
Chicago/Turabian StyleMoin, Afrasim, Bader Huwaimel, Ahmed Alobaida, Mohammed Khaled Bin Break, Danish Iqbal, Rahamat Unissa, Qazi Mohammad Sajid Jamal, Talib Hussain, Dinesh C. Sharma, and Syed Mohd Danish Rizvi. 2022. "Dithymoquinone Analogues as Potential Candidate(s) for Neurological Manifestation Associated with COVID-19: A Therapeutic Strategy for Neuro-COVID" Life 12, no. 7: 1076. https://doi.org/10.3390/life12071076
APA StyleMoin, A., Huwaimel, B., Alobaida, A., Break, M. K. B., Iqbal, D., Unissa, R., Jamal, Q. M. S., Hussain, T., Sharma, D. C., & Rizvi, S. M. D. (2022). Dithymoquinone Analogues as Potential Candidate(s) for Neurological Manifestation Associated with COVID-19: A Therapeutic Strategy for Neuro-COVID. Life, 12(7), 1076. https://doi.org/10.3390/life12071076