
Survey of the Intermolecular Disulfide Bonds Observed in Protein Crystal Structures Deposited in the Protein Data Bank


Abstract
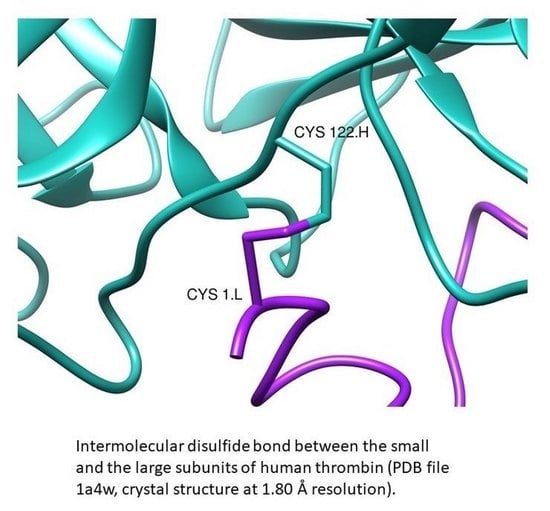
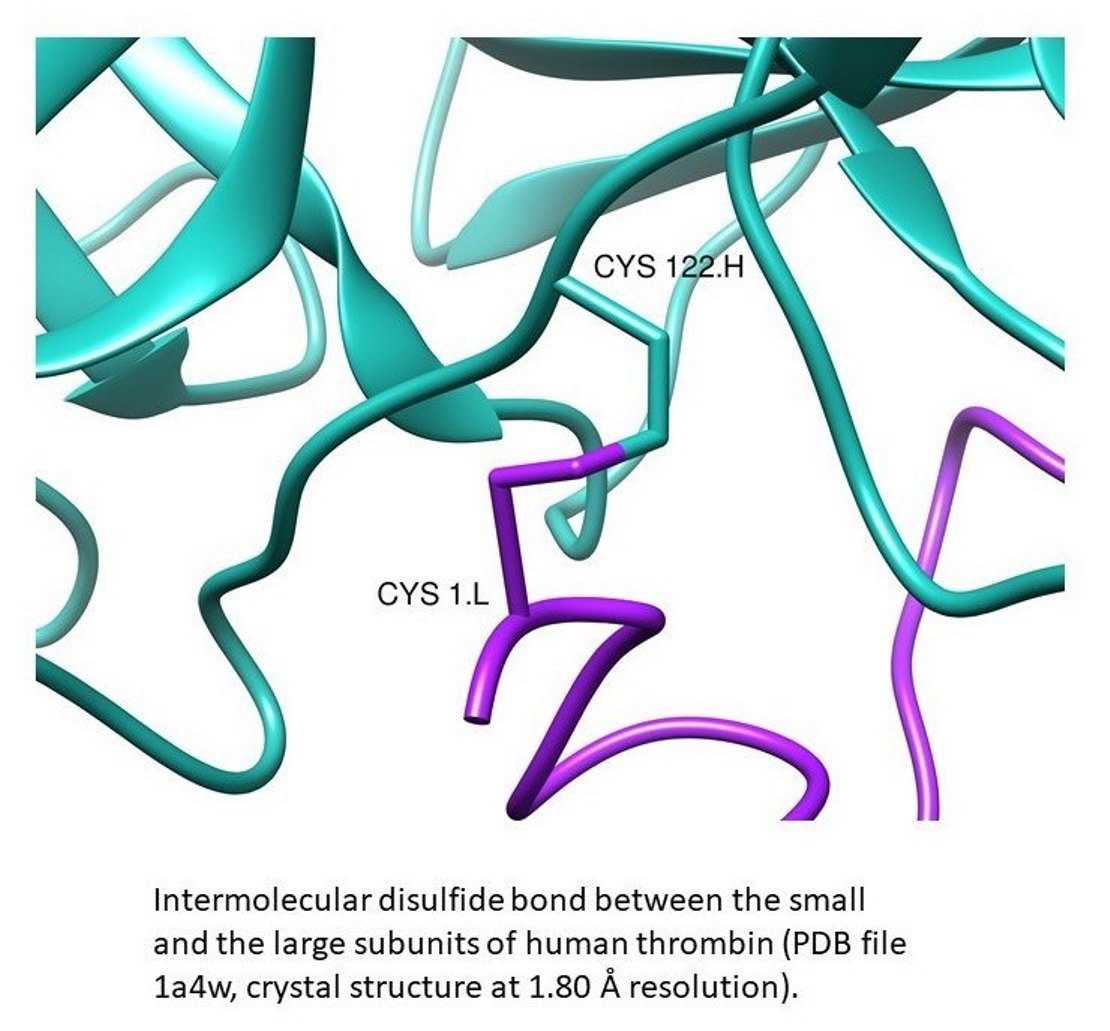
:
1. Introduction
2. Methods
2.1. Data Selection
2.2. DB Identification
2.3. Miscellaneous
3. Results and Discussion
3.1. General Features
3.1.1. How Frequent Are Intermolecular DBs?
3.1.2. Homomeric and Heteromeric Intermolecular DBs
3.1.3. Number of Intermolecular Connections
3.1.4. Amino Acid Composition
3.2. Local Features
3.2.1. Secondary Structures
3.2.2. Solvent Accessibility
3.2.3. Stereochemistry
3.2.4. B-Factors
3.3. Global Features
3.3.1. Enzyme Types
3.3.2. Structural Classes
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mirsky, A.E.; Anson, M.L. Sulfhydryl and disulfide groups of proteins: I. Methods of estimation. J. Gen. Physiol. 1935, 18, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Goldberger, R.; Epstein, C.; Anfinsen, C. Acceleration of Reactivation of Reduced Bovine Pancreatic Ribonuclease by a Microsomal System from. J. Biol. Chem. 1963, 238, 628–635. [Google Scholar] [CrossRef]
- Venetianer, P.; Straub, F. Enzymic Reactivation of Reduced Ribonuclease. Biochim. Biophys. Acta 1963, 67, 166–168. [Google Scholar] [CrossRef]
- Bardwell, J.C.; McGovern, K.; Beckwith, J. Identification of a protein required for disulfide bond formation in vivo. Cell 1991, 67, 581–589. [Google Scholar] [CrossRef]
- Thornton, J.M. Disulphide bridges in globular proteins. J. Mol. Biol. 1981, 151, 261–287. [Google Scholar] [CrossRef]
- Narayan, M. The formation of native disulfide bonds: Treading a fine line in protein folding. Protein J. 2021, 40, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Arai, K.; Iwaoka, M. Flexible folding: Disulfide-containing peptides and proteins choose the pathway depending on the environments. Molecules 2021, 26, 195. [Google Scholar] [CrossRef]
- Fu, J.; Gao, J.; Liang, Z.; Yang, D. PDI-Regulated Disulfide Bond Formation in Protein Folding and Biomolecular Assembly. Molecules 2020, 26, 171. [Google Scholar] [CrossRef]
- Craik, D.J. The folding of disulfide-rich proteins. Antioxid. Redox Signal. 2011, 14, 61–64. [Google Scholar] [CrossRef] [Green Version]
- Hatahet, F.; Boyd, D.; Beckwith, J. Disulfide Bond Formation in Prokaryotes: History, Diversity and Design. Biochim. Biophys. Acta 2014, 1844, 1402–1414. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, J.M.; Riemer, J. Mitochondrial disulfide relay: Redox-regulated protein import into the intermembrane space. J. Biol. Chem. 2012, 287, 4426–4433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feige, M.J.; Hendershot, L.M. Disulfide bonds in ER protein folding and homeostasis. Curr. Opin. Cell Biol. 2011, 23, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, K. Structural basis of protein disulfide bond generation in the cell. Genes Cells 2010, 15, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, M.V.; Laurence, J.S.; Siahaan, T.J. The role of thiols and disulfides on protein stability. Curr. Protein Pept. Sci. 2009, 10, 614–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carugo, O.; Cemazar, M.; Zahariev, S.; Hudaky, I.; Gaspari, Z.; Perczel, A.; Pongor, S. Vicinal disulfide turns. Protein Eng. 2003, 16, 637–639. [Google Scholar] [CrossRef] [Green Version]
- Engelberg, Y.; Ragonis-Bachar, P.; Landau, M. Rare by Natural Selection: Disulfide-Bonded Supramolecular Antimicrobial Peptides. Biomacromolecules 2022, 23, 926–936. [Google Scholar] [CrossRef]
- Yang, L.; Liu, Y.; Wang, N.; Wang, H.; Wang, K.; Luo, X.-L.; Dai, R.-X.; Tao, R.-J.; Wang, H.-J.; Yang, J.-W.; et al. Albumin-based ll37 peptide nanoparticles as a sustained release system against pseudomonas aeruginosa lung infection. ACS Biomater. Sci. Eng. 2021, 7, 1817–1826. [Google Scholar] [CrossRef]
- Liao, X.; Li, H.; Guo, Y.; Yang, F.; Chen, Y.; He, X.; Li, H.; Xia, W.; Mao, Z.-W.; Sun, H. Regulation of DNA-binding activity of the Staphylococcus aureus catabolite control protein A by copper (II)-mediated oxidation. J. Biol. Chem. 2022, 298, 101587. [Google Scholar] [CrossRef]
- Mitra, A.; Sarkar, N. The role of intra and inter-molecular disulfide bonds in modulating amyloidogenesis: A review. Arch. Biochem. Biophys. 2022, 716, 109113. [Google Scholar] [CrossRef]
- Thorn, D.C.; Bahraminejad, E.; Grosas, A.B.; Koudelka, T.; Hoffmann, P.; Mata, J.P.; Devlin, G.L.; Sunde, M.; Ecroyd, H.; Holt, C.; et al. Native disulphide-linked dimers facilitate amyloid fibril formation by bovine milk α S2-casein. Biophys. Chem. 2021, 270, 106530. [Google Scholar] [CrossRef]
- Werner, T.E.R.; Bernson, D.; Esbjörner, E.K.; Rocha, S.; Wittung-Stafshede, P. Amyloid formation of fish β-parvalbumin involves primary nucleation triggered by disulfide-bridged protein dimers. Proc. Natl. Acad. Sci. USA 2020, 117, 27997–28004. [Google Scholar] [CrossRef] [PubMed]
- Fonin, A.V.; Silonov, S.A.; Shpironok, O.G.; Antifeeva, I.A.; Petukhov, A.V.; Romanovich, A.E.; Kuznetsova, I.M.; Uversky, V.N.; Turoverov, K.K. The Role of Non-Specific Interactions in Canonical and ALT-Associated PML-Bodies Formation and Dynamics. Int. J. Mol. Sci. 2021, 22, 5821. [Google Scholar] [CrossRef] [PubMed]
- Martina, J.A.; Guerrero-Gómez, D.; Gómez-Orte, E.; Bárcena, J.A.; Cabello, J.; Miranda-Vizuete, A.; Puertollano, R.A. Conserved cysteine-based redox mechanism sustains TFEB/HLH-30 activity under persistent stress. EMBO J. 2021, 40, e10579. [Google Scholar] [CrossRef] [PubMed]
- Flores-Solis, D.; Mendoza, A.; Rentería-González, I.; Casados-Vazquez, L.E.; Trasviña-Arenas, C.H.; Jiménez-Sandoval, P.; Benítez-Cardoza, C.G.; Río-Portilla, F.d.; Brieba, L.G. Solution structure of the inhibitor of cysteine proteases 1 from Entamoeba histolytica reveals a possible auto regulatory mechanism. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140512. [Google Scholar] [CrossRef]
- Khalaj, A.J.; Sterky, F.H.; Sclip, A.; Schwenk, J.; Brunger, A.T.; Fakler, B.; Südhof, T.C. Deorphanizing FAM19A proteins as pan-neurexin ligands with an unusual biosynthetic binding mechanism. J. Cell Biol. 2020, 219, e202004164. [Google Scholar] [CrossRef]
- Stewart, A.N.; Little, H.C.; Clark, D.J.; Zhang, H.; Wong, G.W. Protein modifications critical for myonectin/erythroferrone secretion and oligomer assembly. Biochemistry 2020, 59, 2684–2697. [Google Scholar] [CrossRef]
- Horx, P.; Geyer, A. Comparing the hinge-type mobility of natural and designed intermolecular bi-disulfide domains. Front. Chem. 2020, 8, 25. [Google Scholar] [CrossRef]
- Zulliger, R.; Conley, S.M.; Mwoyosvi, M.L.; Al-Ubaidi, M.R.; Naash, M.I. Oligomerization of Prph2 and Rom1 is essential for photoreceptor outer segment formation. Hum. Mol. Genet. 2018, 27, 3507–3518. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.B.; Meyer, E.F.J.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef]
- Carugo, O. Random sampling of the Protein Data Bank: RaSPDB. Sci. Rep. 2021, 11, 24178. [Google Scholar] [CrossRef] [PubMed]
- Djinovic-Carugo, K.; Carugo, O. Criteria to extract high quality Protein Data Bank subsets for structure users. Methods Mol. Biol. 2016, 1415, 139–152. [Google Scholar]
- Heinig, M.; Frishman, D. STRIDE: A web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004, 32, W500–W502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pettern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.H.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of PDB related databases for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef]
- Hubbard, S.J.; Thornton, J.M. NACCESS, V2.1.1; Department of Biochemistry and Molecular Biology, University College: London, UK, 1993. [Google Scholar]
- Carugo, O. Atomic displacement parameters in structural biology. Amino Acids 2018, 50, 775–786. [Google Scholar] [CrossRef]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: A structural classification of protein database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- Fox, N.K.; Brenner, S.E.; Chandonia, J.-M. SCOPe: Structural Classification of Proteins—extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 2014, 42, D304–D309. [Google Scholar] [CrossRef]
- Carugo, O.; Argos, P. Correlation between side chain mobility and conformation in protein structures. Protein Eng. 1997, 10, 777–787. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FIND | GREP | |||
|---|---|---|---|---|
| NSS | Identical | Different | Identical | Different |
| 1 | 83.9 (1.3) | 77.3 (1.6) | 86.3 (0.8) | 76.5 (1.6) |
| 2 | 15.6 (0.9) | 22.3 (1.7) | 13.1 (0.7) | 23.0 (1.7) |
| 3 | 0.5 (0.4) | 0.5 (0.2) | 0.6 (0.3) | 0.5 (0.2) |
| ss | DSSP | Stride | ||||||
|---|---|---|---|---|---|---|---|---|
| FIND | GREP | FIND | GREP | |||||
| Intra | Inter | Intra | Inter | Intra | Inter | Intra | Inter | |
| HH | 4.5 (0.2) | 9.8 (0.7) | 4.6 (0.2) | 11.3 (0.6) | 6.1 (0.2) | 15.1 (1.2) | 7.3 (0.7) | 11.2 (2.3) |
| HE | 5.2 (0.2) | 0.2 (0.1) | 5.2 (0.2) | 0.2 (0.1) | 5.3 (0.3) | 0.2 (0.1) | 7.3 (0.5) | 1.6 (1.1) |
| EE | 22.7 (0.3) | 15.8 (0.7) | 22.5 (0.2) | 15.2 (0.8) | 23.0 (0.4) | 14.1 (1.2) | 18.2 (0.7) | 7.8 (1.2) |
| CE | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 6.2 (0.2) | 1.2 (0.2) | 5.5 (0.3) | 1.1 (0.3) |
| TH | 1.9 (0.1) | 2.5 (0.4) | 1.9 (0.1) | 2.5 (0.4) | 4.0 (0.1) | 0.8 (0.2) | 5.1 (0.3) | 0.9 (0.4) |
| TE | 2.0 (0.1) | 0.2 (0.1) | 1.9 (0.0) | 0.2 (0.1) | 5.8 (0.2) | 0.6 (0.2) | 5.1 (0.2) | 0.1 (0.1) |
| TT | 0.5 (0.0) | 2.1 (0.3) | 0.5 (0.0) | 2.2 (0.3) | 6.3 (0.1) | 5.0 (0.6) | 7.0 (0.3) | 5.8 (1.0) |
| Protein | FIND | GREP | ||||
|---|---|---|---|---|---|---|
| Both | Intra-Only | Inter-Only | Both | Intra-Only | Inter-Only | |
| Non-enzyme | 57.3 (1.0) | 47.6 (0.3) | 54.2 (2.0) | 57.0 (1.0) | 47.5 (0.3) | 50.9 (1.7) |
| Oxidoreductases | 1.9 (0.2) | 6.3 (0.2) | 9.8 (0.8) | 2.1 (0.2) | 7.0 (0.2) | 13.5 (1.0) |
| Transferases | 0.8 (0.2) | 3.5 (0.2) | 14.8 (0.7) | 1.5 (0.3) | 3.9 (0.2) | 13.7 (0.7) |
| Hydrolases | 39.2 (1.0) | 41.1 (0.5) | 13.5 (1.4) | 38.1 (1.0) | 39.7 (0.5) | 12.4 (1.3) |
| Lyases | 0.7 (0.2) | 0.9 (0.1) | 3.2 (0.5) | 1.2 (0.2) | 1.0 (0.1) | 3.6 (0.7) |
| Isomerases | 0.0 (0.0) | 0.3 (0.0) | 2.5 (0.6) | 0.0 (0.0) | 0.4 (0.0) | 2.4 (0.4) |
| Ligases | 0.1 (0.1) | 0.3 (0.0) | 2.1 (0.6) | 0.2 (0.1) | 0.4 (0.1) | 3.5 (0.6) |
| Translocases | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
| Class | FIND | GREP | % of Folds | % of Domains |
|---|---|---|---|---|
| a | 11.1 (2.3) | 11.1 (2.5) | 28.6 | 15.8 |
| b | 50.3 (3.7) | 50.3 (3.4) | 17.8 | 27.2 |
| c | 16.0 (2.3) | 16.2 (2.3) | 14.6 | 29.9 |
| d | 22.6 (3.5) | 22.4 (3.3) | 39.1 | 27.1 |
| FIND | ||||
|---|---|---|---|---|
| Class | a | b | c | d |
| a | 13.1 (4.7) | 0.0 (0.0) | 0.4 (0.7) | 0.1 (0.4) |
| b | 0.0 (0.0) | 45.2 (3.9) | 0.1 (0.3) | 1.4 (1.3) |
| c | 0.4 (0.7) | 0.1 (0.3) | 16.6 (2.6) | 0.2 (0.8) |
| d | 0.1 (0.4) | 1.4 (1.3) | 0.2 (0.8) | 20.7 (3.0) |
| GREP | ||||
| Class | a | b | c | d |
| a | 12.3 (4.1) | 0.1 (0.2) | 0.3 (0.6) | 0.2 (0.4) |
| b | 0.1 (0.2) | 39.0 (3.1) | 0.1 (0.4) | 1.3 (1.2) |
| c | 0.3 (0.6) | 0.1 (0.4) | 18.7 (2.4) | 0.2 (0.6) |
| d | 0.2 (0.4) | 1.3 (1.2) | 0.2 (0.6) | 25.8 (3.9) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carugo, O. Survey of the Intermolecular Disulfide Bonds Observed in Protein Crystal Structures Deposited in the Protein Data Bank. Life 2022, 12, 986. https://doi.org/10.3390/life12070986
Carugo O. Survey of the Intermolecular Disulfide Bonds Observed in Protein Crystal Structures Deposited in the Protein Data Bank. Life. 2022; 12(7):986. https://doi.org/10.3390/life12070986
Chicago/Turabian StyleCarugo, Oliviero. 2022. "Survey of the Intermolecular Disulfide Bonds Observed in Protein Crystal Structures Deposited in the Protein Data Bank" Life 12, no. 7: 986. https://doi.org/10.3390/life12070986
APA StyleCarugo, O. (2022). Survey of the Intermolecular Disulfide Bonds Observed in Protein Crystal Structures Deposited in the Protein Data Bank. Life, 12(7), 986. https://doi.org/10.3390/life12070986