
Artificial Intelligence in Cryo-Electron Microscopy




Abstract
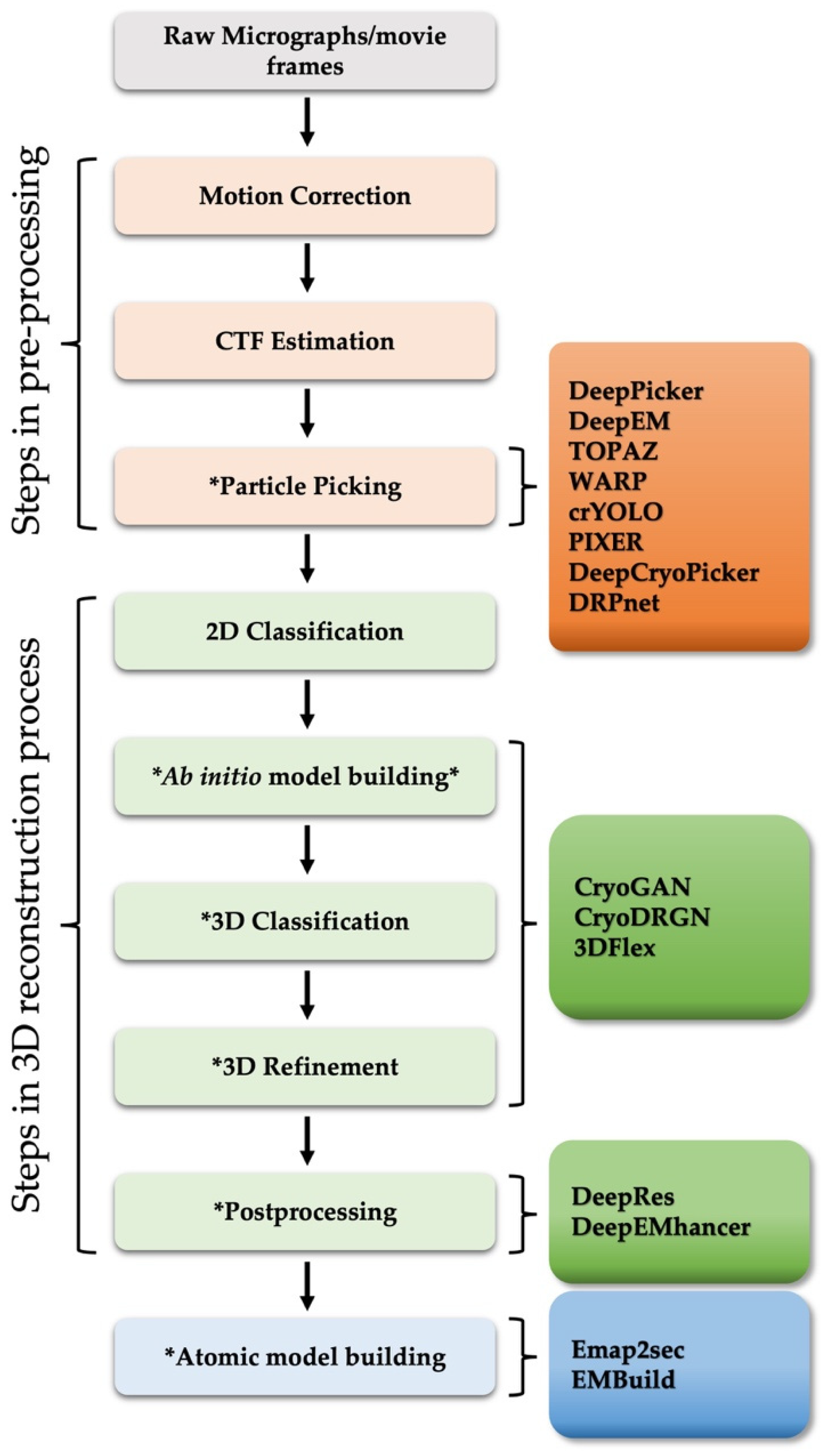
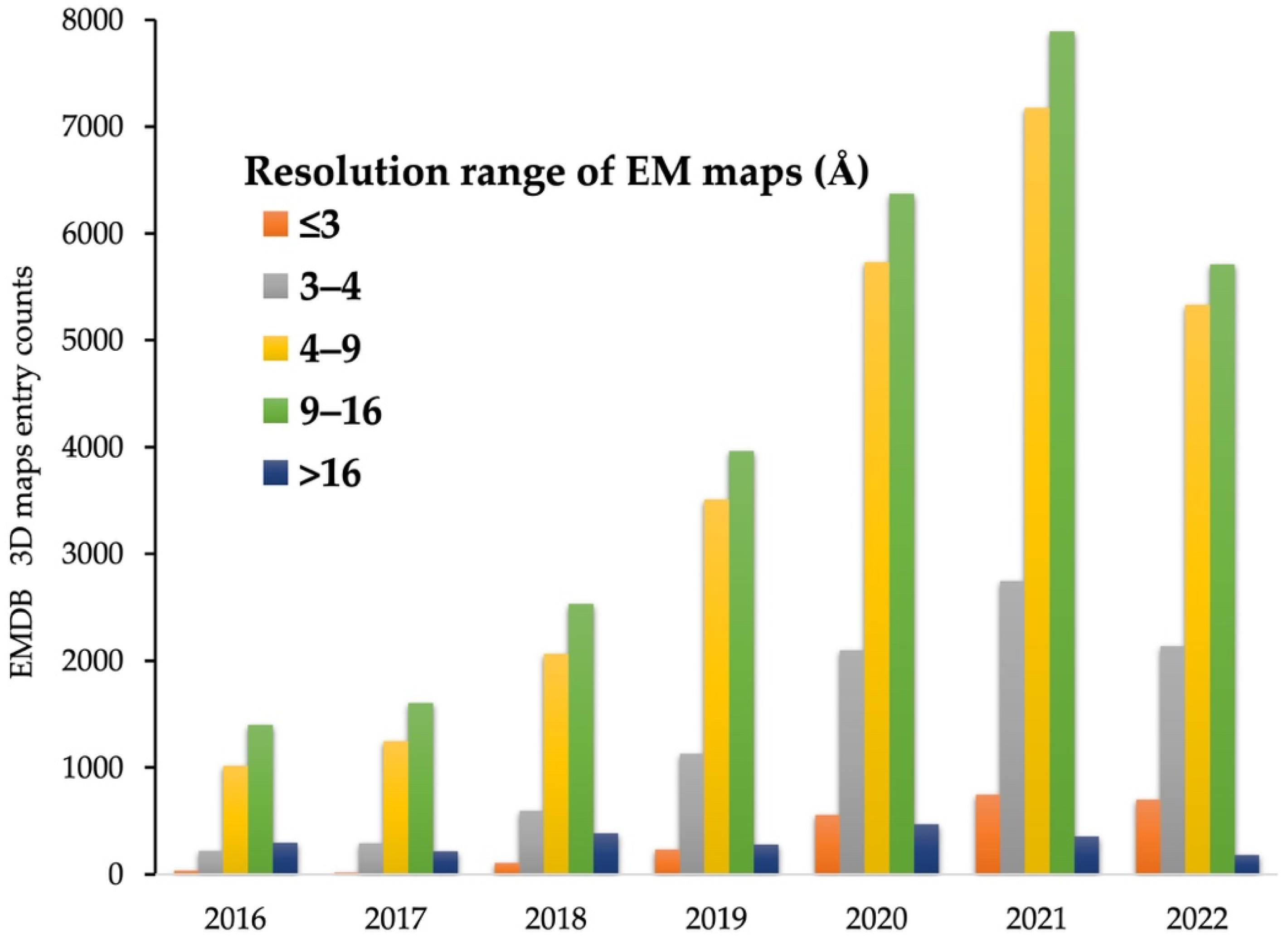
:1. Introduction
2. Pre-Processing: Particle Picking
3. Three-Dimensional (3D) Map Reconstruction
3.1. Model Building, 3D Classification, and 3D Refinement
3.2. Postprocessing
4. Atomic Model Building
5. Future Applications
Author Contributions
Funding
Conflicts of Interest
References
- Berendsen, H.J.; Hayward, S. Collective protein dynamics in relation to function. Curr. Opin. Struct. Biol. 2000, 10, 165–169. [Google Scholar] [CrossRef]
- Chung, J.M.; Jung, H.S. Cryo-electron tomography: A tool for in situ structural analysis of macromolecular complexes. Appl. Spectrosc. Rev. 2017, 53, 195–202. [Google Scholar] [CrossRef]
- Kuhlbrandt, W. Biochemistry. The resolution revolution. Science 2014, 343, 1443–1444. [Google Scholar] [CrossRef] [PubMed]
- A celebration of structural biology. Nat. Methods 2021, 18, 427. [CrossRef]
- Passmore, L.A.; Russo, C.J. Specimen Preparation for High-Resolution Cryo-EM. Methods Enzymol. 2016, 579, 51–86. [Google Scholar] [CrossRef] [PubMed]
- Nakane, T.; Kotecha, A.; Sente, A.; McMullan, G.; Masiulis, S.; Brown, P.; Grigoras, I.T.; Malinauskaite, L.; Malinauskas, T.; Miehling, J.; et al. Single-particle cryo-EM at atomic resolution. Nature 2020, 587, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Yip, K.M.; Fischer, N.; Paknia, E.; Chari, A.; Stark, H. Atomic-resolution protein structure determination by cryo-EM. Nature 2020, 587, 157–161. [Google Scholar] [CrossRef]
- Zivanov, J.; Nakane, T.; Scheres, S.H.W. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 2020, 7, 253–267. [Google Scholar] [CrossRef]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; Brubaker, M.A. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. [Google Scholar] [CrossRef]
- Grant, T.; Rohou, A.; Grigorieff, N. cisTEM, user-friendly software for single-particle image processing. eLife 2018, 7, e35383. [Google Scholar] [CrossRef]
- De la Rosa-Trevin, J.M.; Quintana, A.; Del Cano, L.; Zaldivar, A.; Foche, I.; Gutierrez, J.; Gomez-Blanco, J.; Burguet-Castell, J.; Cuenca-Alba, J.; Abrishami, V.; et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J. Struct. Biol. 2016, 195, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H.R. Conformational changes studied by cryo-electron microscopy. Nat. Struct. Biol. 2000, 7, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Skalidis, I.; Kyrilis, F.L.; Tuting, C.; Hamdi, F.; Chojnowski, G.; Kastritis, P.L. Cryo-EM and artificial intelligence visualize endogenous protein community members. Structure 2022, 30, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.L.; Chiu, W. Comparing cryo-EM structures. J. Struct. Biol. 2018, 204, 523–526. [Google Scholar] [CrossRef]
- Li, Y.; Cash, J.N.; Tesmer, J.J.G.; Cianfrocco, M.A. High-Throughput Cryo-EM Enabled by User-Free Preprocessing Routines. Structure 2020, 28, 858–869. [Google Scholar] [CrossRef]
- Wang, F.; Gong, H.; Liu, G.; Li, M.; Yan, C.; Xia, T.; Li, X.; Zeng, J. DeepPicker: A deep learning approach for fully automated particle picking in cryo-EM. J. Struct. Biol. 2016, 195, 325–336. [Google Scholar] [CrossRef]
- Wagner, T.; Merino, F.; Stabrin, M.; Moriya, T.; Antoni, C.; Apelbaum, A.; Hagel, P.; Sitsel, O.; Raisch, T.; Prumbaum, D.; et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol. 2019, 2, 218. [Google Scholar] [CrossRef]
- Nguyen, N.P.; Ersoy, I.; Gotberg, J.; Bunyak, F.; White, T.A. DRPnet: Automated particle picking in cryo-electron micrographs using deep regression. BMC Bioinform. 2021, 22, 55. [Google Scholar] [CrossRef]
- Al-Azzawi, A.; Ouadou, A.; Max, H.; Duan, Y.; Tanner, J.J.; Cheng, J. DeepCryoPicker: Fully automated deep neural network for single protein particle picking in cryo-EM. BMC Bioinform. 2020, 21, 509. [Google Scholar] [CrossRef]
- Zhu, Y.; Ouyang, Q.; Mao, Y. A deep convolutional neural network approach to single-particle recognition in cryo-electron microscopy. BMC Bioinform. 2017, 18, 348. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Z.; Chen, Y.; Han, R.; Liu, Z.; Sun, F.; Zhang, F. PIXER: An automated particle-selection method based on segmentation using a deep neural network. BMC Bioinform. 2019, 20, 41. [Google Scholar] [CrossRef] [PubMed]
- Bepler, T.; Morin, A.; Rapp, M.; Brasch, J.; Shapiro, L.; Noble, A.J.; Berger, B. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 2019, 16, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Tegunov, D.; Cramer, P. Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 2019, 16, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Zhong, E.D.; Bepler, T.; Berger, B.; Davis, J.H. CryoDRGN: Reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 2021, 18, 176–185. [Google Scholar] [CrossRef]
- Gupta, H.; McCann, M.T.; Donati, L.; Unser, M. CryoGAN: A New Reconstruction Paradigm for Single-Particle Cryo-EM Via Deep Adversarial Learning. IEEE Trans. Comput. Imaging 2021, 7, 759–774. [Google Scholar] [CrossRef]
- Punjani, A.; Fleet, D.J. 3D Flexible Refinement: Structure and Motion of Flexible Proteins from Cryo-EM. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ramirez-Aportela, E.; Mota, J.; Conesa, P.; Carazo, J.M.; Sorzano, C.O.S. DeepRes: A new deep-learning- and aspect-based local resolution method for electron-microscopy maps. IUCrJ 2019, 6, 1054–1063. [Google Scholar] [CrossRef]
- Avramov, T.K.; Vyenielo, D.; Gomez-Blanco, J.; Adinarayanan, S.; Vargas, J.; Si, D. Deep Learning for Validating and Estimating Resolution of Cryo-Electron Microscopy Density Maps. Molecules 2019, 24, 1181. [Google Scholar] [CrossRef]
- Sanchez-Garcia, R.; Gomez-Blanco, J.; Cuervo, A.; Carazo, J.M.; Sorzano, C.O.S.; Vargas, J. DeepEMhancer: A deep learning solution for cryo-EM volume post-processing. Commun. Biol. 2021, 4, 874. [Google Scholar] [CrossRef]
- Maddhuri Venkata Subramaniya, S.R.; Terashi, G.; Kihara, D. Protein secondary structure detection in intermediate-resolution cryo-EM maps using deep learning. Nat. Methods 2019, 16, 911–917. [Google Scholar] [CrossRef]
- He, J.; Lin, P.; Chen, J.; Cao, H.; Huang, S.Y. Model building of protein complexes from intermediate-resolution cryo-EM maps with deep learning-guided automatic assembly. Nat. Commun. 2022, 13, 4066. [Google Scholar] [CrossRef]
- Scheres, S.H. Beam-induced motion correction for sub-megadalton cryo-EM particles. eLife 2014, 3, e03665. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Penczek, P.A.; Schroder, R.; Frank, J. Three-dimensional reconstruction with contrast transfer function correction from energy-filtered cryoelectron micrographs: Procedure and application to the 70S Escherichia coli ribosome. J. Struct. Biol. 1997, 118, 197–219. [Google Scholar] [CrossRef] [PubMed]
- Sigworth, F.J. Principles of cryo-EM single-particle image processing. Microscopy 2016, 65, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Carazo, J.M.; Frank, J. Three-dimensional matching of macromolecular structures obtained from electron microscopy: An application to the 70S and 50S E. coli ribosomal particles. Ultramicroscopy 1988, 25, 13–22. [Google Scholar] [CrossRef]
- Scheres, S.H. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012, 180, 519–530. [Google Scholar] [CrossRef]
- Adiga, U.; Baxter, W.T.; Hall, R.J.; Rockel, B.; Rath, B.K.; Frank, J.; Glaeser, R. Particle picking by segmentation: A comparative study with SPIDER-based manual particle picking. J. Struct. Biol. 2005, 152, 211–220. [Google Scholar] [CrossRef]
- Yu, Z.; Bajaj, C. Detecting circular and rectangular particles based on geometric feature detection in electron micrographs. J. Struct. Biol. 2004, 145, 168–180. [Google Scholar] [CrossRef]
- Scheres, S.H. Semi-automated selection of cryo-EM particles in RELION-1.3. J. Struct. Biol. 2015, 189, 114–122. [Google Scholar] [CrossRef]
- Langlois, R.; Pallesen, J.; Ash, J.T.; Nam Ho, D.; Rubinstein, J.L.; Frank, J. Automated particle picking for low-contrast macromolecules in cryo-electron microscopy. J. Struct. Biol. 2014, 186, 1–7. [Google Scholar] [CrossRef]
- Schmidhuber, J. Deep learning in neural networks: An overview. Neural Netw. 2015, 61, 85–117. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep Residual Learning for Image Recognition. In Proceedings of the 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Iudin, A.; Korir, P.K.; Salavert-Torres, J.; Kleywegt, G.J.; Patwardhan, A. EMPIAR: A public archive for raw electron microscopy image data. Nat. Methods 2016, 13, 387–388. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Redmon, J.; Farhadi, A. YOLO9000: Better, Faster, Stronger. In Proceedings of the 2017 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Honolulu, HI, USA, 21–26 July 2017; pp. 6517–6525. [Google Scholar]
- Dubochet, J.; Adrian, M.; Chang, J.J.; Homo, J.C.; Lepault, J.; McDowall, A.W.; Schultz, P. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 1988, 21, 129–228. [Google Scholar] [CrossRef] [PubMed]
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nat. Methods 2016, 13, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Thuman-Commike, P.A. Single particle macromolecular structure determination via electron microscopy. FEBS Lett. 2001, 505, 199–205. [Google Scholar] [CrossRef]
- Penczek, P.A.; Grassucci, R.A.; Frank, J. The ribosome at improved resolution: New techniques for merging and orientation refinement in 3D cryo-electron microscopy of biological particles. Ultramicroscopy 1994, 53, 251–270. [Google Scholar] [CrossRef]
- Sigworth, F.J. A maximum-likelihood approach to single-particle image refinement. J. Struct. Biol. 1998, 122, 328–339. [Google Scholar] [CrossRef]
- Singer, A.; Sigworth, F.J. Computational Methods for Single-Particle Electron Cryomicroscopy. Annu. Rev. Biomed. Data Sci. 2020, 3, 163–190. [Google Scholar] [CrossRef]
- Nashed, Y.S.G.; Poitevin, F.; Gupta, H.; Woollard, G.; Kagan, M.; Yoon, C.H.; Ratner, D. CryoPoseNet: End-to-End Simultaneous Learning of Single-particle Orientation and 3D Map Reconstruction from Cryo-electron Microscopy Data. In Proceedings of the 2021 IEEE/CVF International Conference on Computer Vision Workshops (ICCVW), Montreal, BC, Canada, 11–17 October 2021; pp. 4049–4059. [Google Scholar]
- Bottou, L.; Curtis, F.E.; Nocedal, J. Optimization Methods for Large-Scale Machine Learning. SIAM Rev. 2018, 60, 223–311. [Google Scholar] [CrossRef]
- Bendory, T.; Bartesaghi, A.; Singer, A. Single-particle cryo-electron microscopy: Mathematical theory, computational challenges, and opportunities. IEEE Signal Process. Mag. 2020, 37, 58–76. [Google Scholar] [CrossRef] [PubMed]
- Ourmazd, A. Cryo-EM, XFELs and the structure conundrum in structural biology. Nat. Methods 2019, 16, 941–944. [Google Scholar] [CrossRef] [PubMed]
- Haselbach, D.; Komarov, I.; Agafonov, D.E.; Hartmuth, K.; Graf, B.; Dybkov, O.; Urlaub, H.; Kastner, B.; Luhrmann, R.; Stark, H. Structure and Conformational Dynamics of the Human Spliceosomal B(act) Complex. Cell 2018, 172, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Nakane, T.; Kimanius, D.; Lindahl, E.; Scheres, S.H. Characterisation of molecular motions in cryo-EM single-particle data by multi-body refinement in RELION. eLife 2018, 7, e36861. [Google Scholar] [CrossRef] [PubMed]
- Penczek, P.A.; Kimmel, M.; Spahn, C.M. Identifying conformational states of macromolecules by eigen-analysis of resampled cryo-EM images. Structure 2011, 19, 1582–1590. [Google Scholar] [CrossRef]
- Tagare, H.D.; Kucukelbir, A.; Sigworth, F.J.; Wang, H.; Rao, M. Directly reconstructing principal components of heterogeneous particles from cryo-EM images. J. Struct. Biol. 2015, 191, 245–262. [Google Scholar] [CrossRef]
- Punjani, A.; Fleet, D.J. 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol. 2021, 213, 107702. [Google Scholar] [CrossRef]
- Bepler, T.; Kelley, K.; Noble, A.J.; Berger, B. Topaz-Denoise: General deep denoising models for cryoEM and cryoET. Nat. Commun. 2020, 11, 5208. [Google Scholar] [CrossRef]
- Goodfelllow, I.J.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative Adversarial Networks. arXiv 2014. [Google Scholar] [CrossRef]
- Kingma, D.P.; Welling, M. Auto-Encoding Variational Bayes. arXiv 2013. [Google Scholar] [CrossRef]
- Cardone, G.; Heymann, J.B.; Steven, A.C. One number does not fit all: Mapping local variations in resolution in cryo-EM reconstructions. J. Struct. Biol. 2013, 184, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Kucukelbir, A.; Sigworth, F.J.; Tagare, H.D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 2014, 11, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Saxton, W.O.; Baumeister, W. The correlation averaging of a regularly arranged bacterial cell envelope protein. J. Microsc. 1982, 127, 127–138. [Google Scholar] [CrossRef]
- Banterle, N.; Bui, K.H.; Lemke, E.A.; Beck, M. Fourier ring correlation as a resolution criterion for super-resolution microscopy. J. Struct. Biol. 2013, 183, 363–367. [Google Scholar] [CrossRef]
- Vilas, J.L.; Gomez-Blanco, J.; Conesa, P.; Melero, R.; Miguel de la Rosa-Trevin, J.; Oton, J.; Cuenca, J.; Marabini, R.; Carazo, J.M.; Vargas, J.; et al. MonoRes: Automatic and Accurate Estimation of Local Resolution for Electron Microscopy Maps. Structure 2018, 26, 337–344.e334. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.L.; Patwardhan, A.; Baker, M.L.; Hryc, C.; Garcia, E.S.; Hudson, B.P.; Lagerstedt, I.; Ludtke, S.J.; Pintilie, G.; Sala, R.; et al. EMDataBank unified data resource for 3DEM. Nucleic Acids Res. 2016, 44, D396–D403. [Google Scholar] [CrossRef]
- Patwardhan, A. Trends in the Electron Microscopy Data Bank (EMDB). Acta Crystallogr. D Struct. Biol. 2017, 73, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.W.; Read, R.J.; Adams, P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 61–69. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Frenz, B.; Walls, A.C.; Egelman, E.H.; Veesler, D.; DiMaio, F. RosettaES: A sampling strategy enabling automated interpretation of difficult cryo-EM maps. Nat. Methods 2017, 14, 797–800. [Google Scholar] [CrossRef]
- Terashi, G.; Kihara, D. De novo main-chain modeling for EM maps using MAINMAST. Nat. Commun. 2018, 9, 1618. [Google Scholar] [CrossRef]
- DiMaio, F.; Song, Y.; Li, X.; Brunner, M.J.; Xu, C.; Conticello, V.; Egelman, E.; Marlovits, T.; Cheng, Y.; Baker, D. Atomic-accuracy models from 4.5-A cryo-electron microscopy data with density-guided iterative local refinement. Nat. Methods 2015, 12, 361–365. [Google Scholar] [CrossRef]
- Esquivel-Rodriguez, J.; Kihara, D. Computational methods for constructing protein structure models from 3D electron microscopy maps. J. Struct. Biol. 2013, 184, 93–102. [Google Scholar] [CrossRef]
- Bharat, T.A.; Scheres, S.H. Resolving macromolecular structures from electron cryo-tomography data using subtomogram averaging in RELION. Nat. Protoc. 2016, 11, 2054–2065. [Google Scholar] [CrossRef] [PubMed]
- Castano-Diez, D.; Zanetti, G. In situ structure determination by subtomogram averaging. Curr. Opin. Struct. Biol. 2019, 58, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Schur, F.K. Toward high-resolution in situ structural biology with cryo-electron tomography and subtomogram averaging. Curr Opin. Struct. Biol. 2019, 58, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lasker, K.; Sali, A.; Wolfson, H.J. Determining macromolecular assembly structures by molecular docking and fitting into an electron density map. Proteins 2010, 78, 3205–3211. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, C.P.; Bonvin, M. Fast and sensitive rigid-body fitting into cryo-EM density maps with PowerFit. AIMS Biophys. 2015, 2, 73–87. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, W.; Pearce, R.; Zhang, Y.; Shen, H.B. Fitting Low-Resolution Protein Structures into Cryo-EM Density Maps by Multiobjective Optimization of Global and Local Correlations. J. Phys. Chem. B 2021, 125, 528–538. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkoczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef]
- Langer, G.; Cohen, S.X.; Lamzin, V.S.; Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. [Google Scholar] [CrossRef]
- Zhou, Z.; Siddiquee, M.M.R.; Tajbakhsh, N.; Liang, J. UNet++: Redesigning Skip Connections to Exploit Multiscale Features in Image Segmentation. IEEE Trans. Med. Imaging 2020, 39, 1856–1867. [Google Scholar] [CrossRef]
- Rawat, W.; Wang, Z. Deep Convolutional Neural Networks for Image Classification: A Comprehensive Review. Neural Comput. 2017, 29, 2352–2449. [Google Scholar] [CrossRef] [PubMed]
- Gavin, A.C.; Aloy, P.; Grandi, P.; Krause, R.; Boesche, M.; Marzioch, M.; Rau, C.; Jensen, L.J.; Bastuck, S.; Dumpelfeld, B.; et al. Proteome survey reveals modularity of the yeast cell machinery. Nature 2006, 440, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, P.L.; O’Reilly, F.J.; Bock, T.; Li, Y.; Rogon, M.Z.; Buczak, K.; Romanov, N.; Betts, M.J.; Bui, K.H.; Hagen, W.J.; et al. Capturing protein communities by structural proteomics in a thermophilic eukaryote. Mol. Syst. Biol. 2017, 13, 936. [Google Scholar] [CrossRef] [PubMed]
- Han, B.G.; Dong, M.; Liu, H.; Camp, L.; Geller, J.; Singer, M.; Hazen, T.C.; Choi, M.; Witkowska, H.E.; Ball, D.A.; et al. Survey of large protein complexes in D. vulgaris reveals great structural diversity. Proc. Natl. Acad. Sci. USA 2009, 106, 16580–16585. [Google Scholar] [CrossRef]
- Kyrilis, F.L.; Semchonok, D.A.; Skalidis, I.; Tuting, C.; Hamdi, F.; O’Reilly, F.J.; Rappsilber, J.; Kastritis, P.L. Integrative structure of a 10-megadalton eukaryotic pyruvate dehydrogenase complex from native cell extracts. Cell Rep. 2021, 34, 108727. [Google Scholar] [CrossRef]
- Su, C.C.; Lyu, M.; Morgan, C.E.; Bolla, J.R.; Robinson, C.V.; Yu, E.W. A ‘Build and Retrieve’ methodology to simultaneously solve cryo-EM structures of membrane proteins. Nat. Methods 2021, 18, 69–75. [Google Scholar] [CrossRef]
- Kyrilis, F.L.; Belapure, J.; Kastritis, P.L. Detecting Protein Communities in Native Cell Extracts by Machine Learning: A Structural Biologist’s Perspective. Front. Mol. Biosci. 2021, 8, 660542. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zidek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Name | Application Area | Reference |
|---|---|---|
| DeepPicker | Particle Recognition | Wang et al., 2016 [16] |
| DeepEM | Particle Recognition | Zhu et al., 2017 [20] |
| TOPAZ | Particle Recognition | Bepler et al., 2019 [22] |
| WARP | Particle Recognition | Tegunov et al., 2019 [23] |
| crYOLO | Particle Recognition | Wagner et al., 2019 [17] |
| PIXER | Particle Recognition | Zhang et al., 2019 [21] |
| DeepCryoPicker | Particle Recognition | Al-Azzawi et al., 2020 [19] |
| DRPnet | Particle Recognition | Nguyen et al., 2021 [18] |
| CryoGAN | 3D Reconstruction | Gupta et al., 2021 [25] |
| CryoDRGN | 3D Reconstruction | Zhong et al., 2021 [24] |
| 3DFlex | 3D Reconstruction | Punjani et al., 2021 [26] |
| DeepRes | Local resolution | Ramirez-Aportela et al., 2019 [27] |
| DeepEMhancer | Map Sharpening | Sanchez-Garcia et al., 2021 [29] |
| Emap2sec | Model building | Maddhuri Venkata Subramaniya et al., 2019 [30] |
| EMBuild | Model building | He et al., 2022 [31] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, J.M.; Durie, C.L.; Lee, J. Artificial Intelligence in Cryo-Electron Microscopy. Life 2022, 12, 1267. https://doi.org/10.3390/life12081267
Chung JM, Durie CL, Lee J. Artificial Intelligence in Cryo-Electron Microscopy. Life. 2022; 12(8):1267. https://doi.org/10.3390/life12081267
Chicago/Turabian StyleChung, Jeong Min, Clarissa L. Durie, and Jinseok Lee. 2022. "Artificial Intelligence in Cryo-Electron Microscopy" Life 12, no. 8: 1267. https://doi.org/10.3390/life12081267
APA StyleChung, J. M., Durie, C. L., & Lee, J. (2022). Artificial Intelligence in Cryo-Electron Microscopy. Life, 12(8), 1267. https://doi.org/10.3390/life12081267