
The Immune System as a Therapeutic Target for Alzheimer’s Disease


Abstract
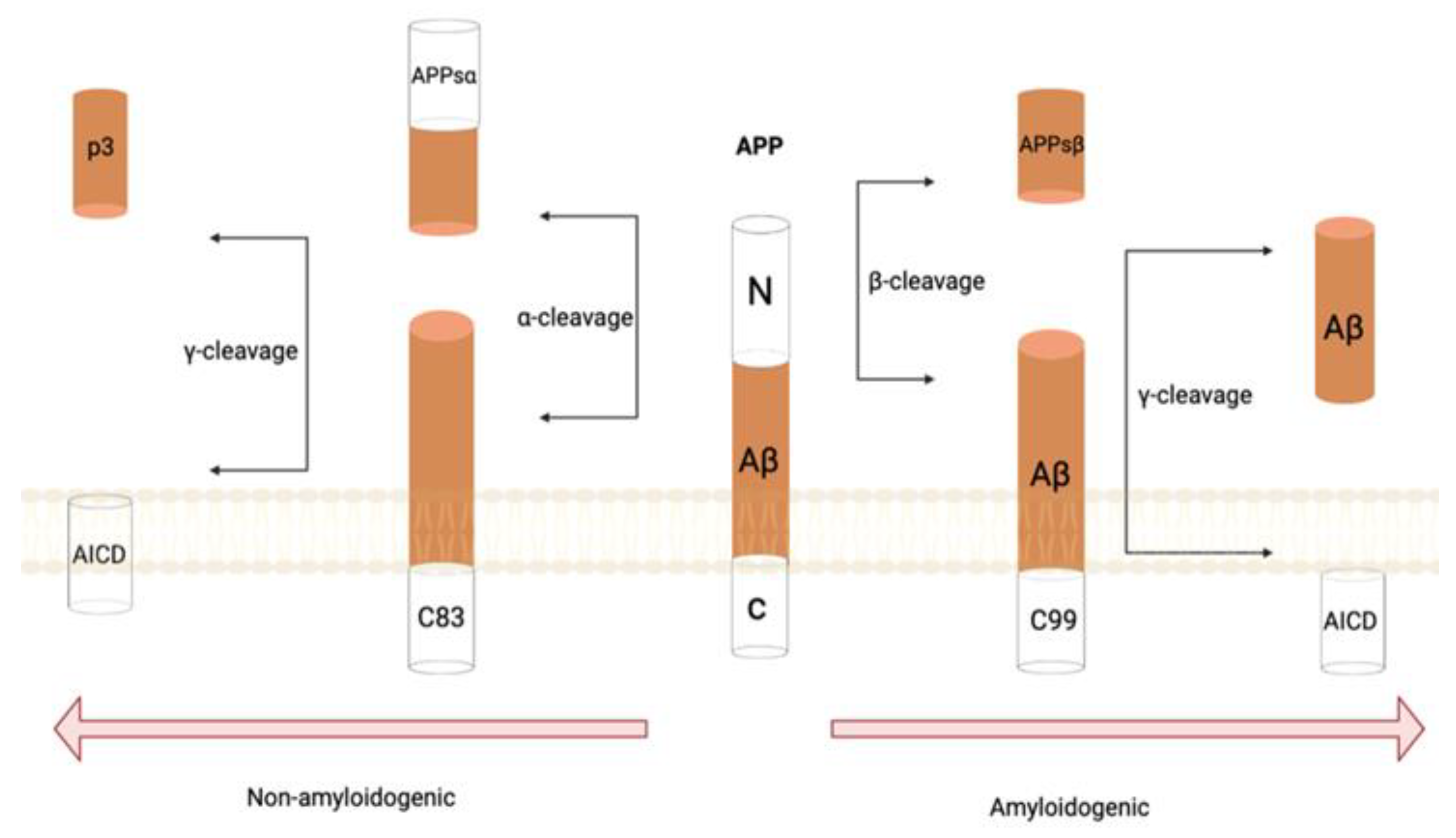
:1. Introduction
2. Pathological Theories of AD
3. Effects of Aging on AD
3.1. Effects of the Peripheral Immune Cells in Aging-Related Brain Homeostasis
3.2. The Innate Immune System
3.3. The Adaptive Immune System
3.4. Central-Peripheral Neuroimmune Crosstalk in AD
4. Risk Profile of AD Patients
5. Immunotherapy
5.1. Active Immunotherapies

{kind=link}
{kind=link}
| Drug Name | Epitope | Mechanism of Action | Composition | Clinical Effects |
|---|---|---|---|---|
| AN1792 [76] | Aβ42 | Reducing the formation of Aβ plaques | Full length human Aβ42 and QS21 adjuvant | Unsafe immune T-cell response resulting in meningoencephalitis |
| CAD106 [144] | Aβ1-6 | Inhibits Aβ aggregation | Shortened beta-amyloid fragments | Safe, immunogenic but no reported clinical efficacy |
| ABVac40 [145,153] | Aβ33-40 | Target the C-terminus of Aβ40 | Aβ33-40, KHL, and alum hydroxide adjuvant | Safe, immunogenic, clinical results study to conclude in February 2022 |
| ACI24 [147,154] | Aβ1-16 | Inhibit the formation of Aβ plaques | Liposome based | Ongoing trial to measure safety, immunogenicity and tolerability |
| UB-311 [155,156] | Aβ1-14 | Induce Aβ antibody response | Aβ1-14 and linked to a helper T-cell peptide epitope | Safe, immunogenic, and tolerable; but efficacy data are not published |
| Peptide vaccine V950 [150,157] | Aβ | Produce Aβ antibodies | Aβ1-14 conjugated to ISCOMATRIX | Terminated study with no clinical data |
| ACC-001 [158,159] | Aβ1-7 | Reducing the formation of Aβ plaques | Aβ1-7, non-toxic form of diphtheria toxin, QS-21 adjuvant | Trial terminated due to no improvement in cognition |
| LU AF20513 [152,160] | Aβ1-12 | Activate B cell polyclonal antibodies | Aβ1-12 and tetanus toxin | Terminated due to efficacy of vaccine |
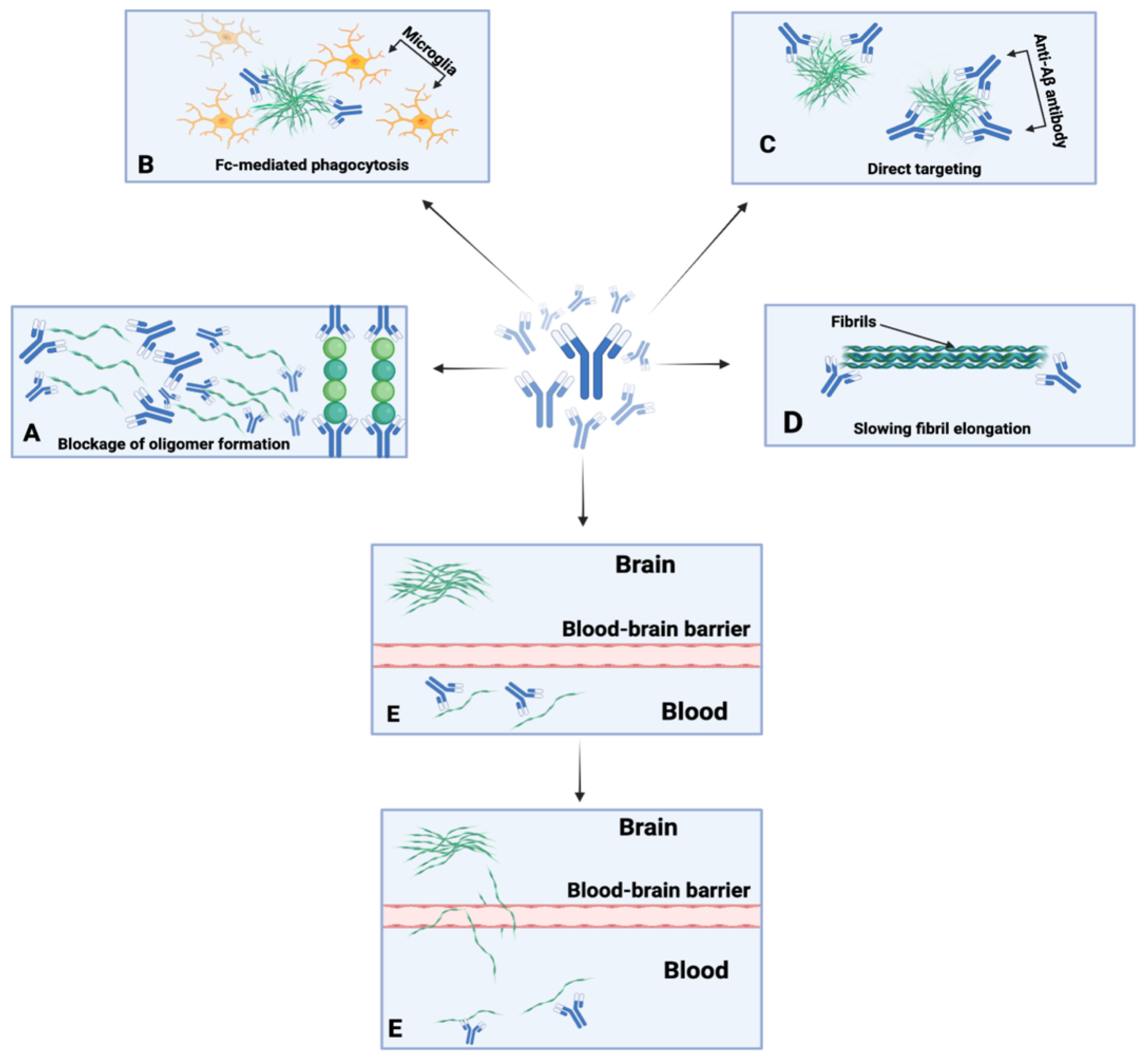
5.2. Passive Immunotherapies
| Drug Name | Mechanism of Action | Clinical Effects |
|---|---|---|
| Bapineuzumab [183,184] | Target Aβ plaques and induce Fc-receptor-mediated phagocytosis | No clinical benefit and ARIA was observed resulting in termination |
| Gantenerumab [165,185] | Inhibits formation of Aβ plaques | No clinical benefit |
| Crenezumab [168] | Inhibits Aβ aggregation and assists with disaggregation | Ongoing trial determining efficacy of treatment |
| Solanezumab [186,187] | Targets the mid-domain of soluble Aβ | No clinical efficacy reported |
| Aducanumab [178,188] | Reduce Aβ oligomers | Phase III trials terminated due to futility analysis (no cognitive benefits), received approval for medical use by FDA |
| BAN2401 [182] | Reduces large, soluble Aβ protofibrils | Safe, tolerable, and unknown efficacy trial will run until October 2027 |
6. Future Direction of AD Immunotherapy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schachter, A.S.; Davis, K.L. Alzheimer’s disease. Dialogues Clin. Neurosci. 2000, 2, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2014, 76 Pt A, 27–50. [Google Scholar] [CrossRef]
- Bishop, G.M.; Robinson, S.R. Physiological roles of amyloid-beta and implications for its removal in Alzheimer’s disease. Drugs Aging 2004, 21, 621–630. [Google Scholar] [CrossRef]
- Jellinger, K.A. Pathobiological Subtypes of Alzheimer Disease. Dement Geriatr. Cogn. Disord. 2020, 49, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.; Verhagen, C.; Hernández-Cabrera, J.A.; Cavallin, L.; Guo, C.J.; Ekman, U.; Muehlboeck, J.S.; Simmons, A.; Barroso, J.; Wahlund, L.O.; et al. Distinct subtypes of Alzheimer’s disease based on patterns of brain atrophy: Longitudinal trajectories and clinical applications. Sci. Rep. 2017, 7, 46263. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Jiang, Q.; McDermott, J.; Han, J.J. Aging and Alzheimer’s disease: Comparison and associations from molecular to system level. Aging Cell 2018, 17, e12802. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.; Wahlund, L.O.; Westman, E. The heterogeneity within Alzheimer’s disease. Aging 2018, 10, 3058–3060. [Google Scholar] [CrossRef]
- Birkenbihl, C.; Salimi, Y.; Fröhlich, H.; Japanese Alzheimer’s Disease Neuroimaging Initiative; Alzheimer’s Disease Neuroimaging Initiative. Unraveling the heterogeneity in Alzheimer’s disease progression across multiple cohorts and the implications for data-driven disease modeling. Alzheimer’s Dement 2021, 18, 251–261. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement 2018, 14, 535–562. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, A.; Condello, C.; Stöhr, J.; Yue, W.; Rivera, B.M.; Lee, J.C.; Woerman, A.L.; Halliday, G.; van Duinen, S.; Ingelsson, M.; et al. Aβ and tau prion-like activities decline with longevity in the Alzheimer’s disease human brain. Sci. Transl. Med. 2019, 11, eaat8462. [Google Scholar] [CrossRef] [PubMed]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed]
- De Simone, A.; Naldi, M.; Tedesco, D.; Bartolini, M.; Davani, L.; Andrisano, V. Advanced analytical methodologies in Alzheimer’s disease drug discovery. J. Pharm. Biomed. Anal. 2020, 178, 112899. [Google Scholar] [CrossRef]
- Huang, X. Alzheimer’s Disease: Drug Discovery; Exon Publications: Brisbane, Australia, 2020. [Google Scholar]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef]
- Boche, D.; Nicoll, J.A.R. Invited Review—Understanding cause and effect in Alzheimer’s pathophysiology: Implications for clinical trials. Neuropathol. Appl. Neurobiol. 2020, 46, 623–640. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Gendreau, K.L.; Hall, G.F. Tangles, Toxicity, and Tau Secretion in AD—New Approaches to a Vexing Problem. Front. Neurol. 2013, 4, 160. [Google Scholar] [CrossRef]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Paudel, Y.N.; Papageorgiou, S.G.; Piperi, C. APOE Genotype and Alzheimer’s Disease: The Influence of Lifestyle and Environmental Factors. ACS Chem. Neurosci. 2021, 12, 2749–2764. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Guerreiro, R.; Bras, J. The age factor in Alzheimer’s disease. Genome Med. 2015, 7, 106. [Google Scholar] [CrossRef]
- Pinti, M.; Appay, V.; Campisi, J.; Frasca, D.; Fülöp, T.; Sauce, D.; Larbi, A.; Weinberger, B.; Cossarizza, A. Aging of the immune system: Focus on inflammation and vaccination. Eur. J. Immunol. 2016, 46, 2286–2301. [Google Scholar] [CrossRef]
- Cao, W.; Zheng, H. Peripheral immune system in aging and Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 51. [Google Scholar] [CrossRef]
- Goldberg, E.L.; Dixit, V.D. Drivers of age-related inflammation and strategies for healthspan extension. Immunol. Rev. 2015, 265, 63–74. [Google Scholar] [CrossRef]
- Netea, M.G.; van de Veerdonk, F.L.; van der Meer, J.W.; Dinarello, C.A.; Joosten, L.A. Inflammasome-independent regulation of IL-1-family cytokines. Annu. Rev. Immunol. 2015, 33, 49–77. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Chang, J.; Lartigue, L.; Bolen, C.R.; Haddad, F.; Gaudilliere, B.; Ganio, E.A.; Fragiadakis, G.K.; Spitzer, M.H.; Douchet, I.; et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat. Med. 2017, 23, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.B.; Sanders, J.L.; Kizer, J.R.; Boudreau, R.M.; Odden, M.C.; Zeki Al Hazzouri, A.; Arnold, A.M. Trajectories of function and biomarkers with age: The CHS All Stars Study. Int. J. Epidemiol. 2016, 45, 1135–1145. [Google Scholar] [CrossRef]
- Martinez-Jimenez, C.P.; Eling, N.; Chen, H.C.; Vallejos, C.A.; Kolodziejczyk, A.A.; Connor, F.; Stojic, L.; Rayner, T.F.; Stubbington, M.J.T.; Teichmann, S.A.; et al. Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science 2017, 355, 1433–1436. [Google Scholar] [CrossRef]
- Enge, M.; Arda, H.E.; Mignardi, M.; Beausang, J.; Bottino, R.; Kim, S.K.; Quake, S.R. Single-Cell Analysis of Human Pancreas Reveals Transcriptional Signatures of Aging and Somatic Mutation Patterns. Cell 2017, 171, 321–330.e14. [Google Scholar] [CrossRef]
- Cheung, P.; Vallania, F.; Warsinske, H.C.; Donato, M.; Schaffert, S.; Chang, S.E.; Dvorak, M.; Dekker, C.L.; Davis, M.M.; Utz, P.J.; et al. Single-Cell Chromatin Modification Profiling Reveals Increased Epigenetic Variations with Aging. Cell 2018, 173, 1385–1397.e14. [Google Scholar] [CrossRef]
- Nikolich-Žugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 10–19. [Google Scholar] [CrossRef]
- Ucar, D.; Márquez, E.J.; Chung, C.H.; Marches, R.; Rossi, R.J.; Uyar, A.; Wu, T.C.; George, J.; Stitzel, M.L.; Palucka, A.K.; et al. The chromatin accessibility signature of human immune aging stems from CD8. J. Exp. Med. 2017, 214, 3123–3144. [Google Scholar] [CrossRef]
- Chiu, B.C.; Martin, B.E.; Stolberg, V.R.; Chensue, S.W. Cutting edge: Central memory CD8 T cells in aged mice are virtual memory cells. J. Immunol. 2013, 191, 5793–5796. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef]
- Villeda, S.A.; Luo, J.; Mosher, K.I.; Zou, B.; Britschgi, M.; Bieri, G.; Stan, T.M.; Fainberg, N.; Ding, Z.; Eggel, A.; et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011, 477, 90–94. [Google Scholar] [CrossRef]
- Sinha, M.; Jang, Y.C.; Oh, J.; Khong, D.; Wu, E.Y.; Manohar, R.; Miller, C.; Regalado, S.G.; Loffredo, F.S.; Pancoast, J.R.; et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science 2014, 344, 649–652. [Google Scholar] [CrossRef]
- Middeldorp, J.; Lehallier, B.; Villeda, S.A.; Miedema, S.S.; Evans, E.; Czirr, E.; Zhang, H.; Luo, J.; Stan, T.; Mosher, K.I.; et al. Preclinical Assessment of Young Blood Plasma for Alzheimer Disease. JAMA Neurol. 2016, 73, 1325–1333. [Google Scholar] [CrossRef]
- Castellano, J.M.; Mosher, K.I.; Abbey, R.J.; McBride, A.A.; James, M.L.; Berdnik, D.; Shen, J.C.; Zou, B.; Xie, X.S.; Tingle, M.; et al. Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nature 2017, 544, 488–492. [Google Scholar] [CrossRef]
- Groh, N.; Bühler, A.; Huang, C.; Li, K.W.; van Nierop, P.; Smit, A.B.; Fändrich, M.; Baumann, F.; David, D.C. Age-Dependent Protein Aggregation Initiates Amyloid-β Aggregation. Front. Aging Neurosci. 2017, 9, 138. [Google Scholar] [CrossRef] [Green Version]
- Tomiyama, T.; Matsuyama, S.; Iso, H.; Umeda, T.; Takuma, H.; Ohnishi, K.; Ishibashi, K.; Teraoka, R.; Sakama, N.; Yamashita, T.; et al. A mouse model of amyloid beta oligomers: Their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J. Neurosci. 2010, 30, 4845–4856. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Korin, B.; Ben-Shaanan, T.L.; Schiller, M.; Dubovik, T.; Azulay-Debby, H.; Boshnak, N.T.; Koren, T.; Rolls, A. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat. Neurosci. 2017, 20, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Kipnis, J.; Cohen, H.; Cardon, M.; Ziv, Y.; Schwartz, M. T cell deficiency leads to cognitive dysfunction: Implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc. Natl. Acad. Sci. USA 2004, 101, 8180–8185. [Google Scholar] [CrossRef] [PubMed]
- Ziv, Y.; Ron, N.; Butovsky, O.; Landa, G.; Sudai, E.; Greenberg, N.; Cohen, H.; Kipnis, J.; Schwartz, M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat. Neurosci. 2006, 9, 268–275. [Google Scholar] [CrossRef]
- Filiano, A.J.; Xu, Y.; Tustison, N.J.; Marsh, R.L.; Baker, W.; Smirnov, I.; Overall, C.C.; Gadani, S.P.; Turner, S.D.; Weng, Z.; et al. Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature 2016, 535, 425–429. [Google Scholar] [CrossRef]
- Derecki, N.C.; Cardani, A.N.; Yang, C.H.; Quinnies, K.M.; Crihfield, A.; Lynch, K.R.; Kipnis, J. Regulation of learning and memory by meningeal immunity: A key role for IL-4. J. Exp. Med. 2010, 207, 1067–1080. [Google Scholar] [CrossRef]
- Baruch, K.; Deczkowska, A.; David, E.; Castellano, J.M.; Miller, O.; Kertser, A.; Berkutzki, T.; Barnett-Itzhaki, Z.; Bezalel, D.; Wyss-Coray, T.; et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 2014, 346, 89–93. [Google Scholar] [CrossRef]
- Balusu, S.; Brkic, M.; Libert, C.; Vandenbroucke, R.E. The choroid plexus-cerebrospinal fluid interface in Alzheimer’s disease: More than just a barrier. Neural Regen. Res. 2016, 11, 534–537. [Google Scholar] [CrossRef]
- Baruch, K.; Ron-Harel, N.; Gal, H.; Deczkowska, A.; Shifrut, E.; Ndifon, W.; Mirlas-Neisberg, N.; Cardon, M.; Vaknin, I.; Cahalon, L.; et al. CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proc. Natl. Acad. Sci. USA 2013, 110, 2264–2269. [Google Scholar] [CrossRef] [Green Version]
- Deczkowska, A.; Baruch, K.; Schwartz, M. Type I/II Interferon Balance in the Regulation of Brain Physiology and Pathology. Trends Immunol. 2016, 37, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Montecino-Rodriguez, E.; Berent-Maoz, B.; Dorshkind, K. Causes, consequences, and reversal of immune system aging. J. Clin. Investig. 2013, 123, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef]
- Cronk, J.C.; Filiano, A.J.; Louveau, A.; Marin, I.; Marsh, R.; Ji, E.; Goldman, D.H.; Smirnov, I.; Geraci, N.; Acton, S.; et al. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J. Exp. Med. 2018, 215, 1627–1647. [Google Scholar] [CrossRef]
- Butovsky, O.; Kunis, G.; Koronyo-Hamaoui, M.; Schwartz, M. Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer’s disease model. Eur. J. Neurosci. 2007, 26, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, J.D.; Ulland, T.K.; Colonna, M.; Holtzman, D.M. Elucidating the Role of TREM2 in Alzheimer’s Disease. Neuron 2017, 94, 237–248. [Google Scholar] [CrossRef]
- Mori, Y.; Yoshino, Y.; Ochi, S.; Yamazaki, K.; Kawabe, K.; Abe, M.; Kitano, T.; Ozaki, Y.; Yoshida, T.; Numata, S.; et al. TREM2 mRNA Expression in Leukocytes Is Increased in Alzheimer’s Disease and Schizophrenia. PLoS ONE 2015, 10, e0136835. [Google Scholar] [CrossRef]
- Tan, Y.J.; Ng, A.S.L.; Vipin, A.; Lim, J.K.W.; Chander, R.J.; Ji, F.; Qiu, Y.; Ting, S.K.S.; Hameed, S.; Lee, T.S.; et al. Higher Peripheral TREM2 mRNA Levels Relate to Cognitive Deficits and Hippocampal Atrophy in Alzheimer’s Disease and Amnestic Mild Cognitive Impairment. J. Alzheimer’s Dis. 2017, 58, 413–423. [Google Scholar] [CrossRef]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef]
- Gabbita, S.P.; Johnson, M.F.; Kobritz, N.; Eslami, P.; Poteshkina, A.; Varadarajan, S.; Turman, J.; Zemlan, F.; Harris-White, M.E. Oral TNFα Modulation Alters Neutrophil Infiltration, Improves Cognition and Diminishes Tau and Amyloid Pathology in the 3xTgAD Mouse Model. PLoS ONE 2015, 10, e0137305. [Google Scholar] [CrossRef]
- Gaskin, F.; Finley, J.; Fang, Q.; Xu, S.; Fu, S.M. Human antibodies reactive with beta-amyloid protein in Alzheimer’s disease. J. Exp. Med. 1993, 177, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Dodel, R.; Hampel, H.; Buerger, K.; Lin, S.; Eastwood, B.; Bales, K.; Gao, F.; Moeller, H.J.; Oertel, W.; et al. Reduced levels of amyloid beta-peptide antibody in Alzheimer disease. Neurology 2001, 57, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Hall, E.; Tuzova, M.; Dobbs, M.; Jons, M.; Anderson, C.; Woodward, J.; Guo, Z.; Fu, W.; Kryscio, R.; et al. Autoantibodies to amyloid beta-peptide (Abeta) are increased in Alzheimer’s disease patients and Abeta antibodies can enhance Abeta neurotoxicity: Implications for disease pathogenesis and vaccine development. Neuromol. Med. 2003, 3, 29–39. [Google Scholar] [CrossRef]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef]
- Gilman, S.; Koller, M.; Black, R.S.; Jenkins, L.; Griffith, S.G.; Fox, N.C.; Eisner, L.; Kirby, L.; Rovira, M.B.; Forette, F.; et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005, 64, 1553–1562. [Google Scholar] [CrossRef]
- Foroutan, N.; Hopkins, R.B.; Tarride, J.E.; Florez, I.D.; Levine, M. Safety and efficacy of active and passive immunotherapy in mild-to-moderate Alzheimer’s disease: A systematic review and network meta-analysis. Clin. Investig. Med. 2019, 42, E53–E65. [Google Scholar] [CrossRef]
- Graham, W.V.; Bonito-Oliva, A.; Sakmar, T.P. Update on Alzheimer’s Disease Therapy and Prevention Strategies. Annu. Rev. Med. 2017, 68, 413–430. [Google Scholar] [CrossRef]
- Lemere, C.A.; Masliah, E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat. Rev. Neurol. 2010, 6, 108–119. [Google Scholar] [CrossRef]
- Acharya, N.K.; Nagele, E.P.; Han, M.; Nagele, R.G. Autoantibodies: Double agents in human disease. Sci. Transl. Med. 2013, 5, 186fs119. [Google Scholar] [CrossRef]
- Nagele, E.P.; Han, M.; Acharya, N.K.; DeMarshall, C.; Kosciuk, M.C.; Nagele, R.G. Natural IgG autoantibodies are abundant and ubiquitous in human sera, and their number is influenced by age, gender, and disease. PLoS ONE 2013, 8, e60726. [Google Scholar] [CrossRef] [Green Version]
- DeMarshall, C.A.; Nagele, E.P.; Sarkar, A.; Acharya, N.K.; Godsey, G.; Goldwaser, E.L.; Kosciuk, M.; Thayasivam, U.; Han, M.; Belinka, B.; et al. Detection of Alzheimer’s disease at mild cognitive impairment and disease progression using autoantibodies as blood-based biomarkers. Alzheimer’s Dement 2016, 3, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Marsh, S.E.; Abud, E.M.; Lakatos, A.; Karimzadeh, A.; Yeung, S.T.; Davtyan, H.; Fote, G.M.; Lau, L.; Weinger, J.G.; Lane, T.E.; et al. The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc. Natl. Acad. Sci. USA 2016, 113, E1316–E1325. [Google Scholar] [CrossRef] [PubMed]
- Relkin, N.R.; Thomas, R.G.; Rissman, R.A.; Brewer, J.B.; Rafii, M.S.; van Dyck, C.H.; Jack, C.R.; Sano, M.; Knopman, D.S.; Raman, R.; et al. A phase 3 trial of IV immunoglobulin for Alzheimer disease. Neurology 2017, 88, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Togo, T.; Akiyama, H.; Iseki, E.; Kondo, H.; Ikeda, K.; Kato, M.; Oda, T.; Tsuchiya, K.; Kosaka, K. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J. Neuroimmunol. 2002, 124, 83–92. [Google Scholar] [CrossRef]
- Itagaki, S.; McGeer, P.L.; Akiyama, H. Presence of T-cytotoxic suppressor and leucocyte common antigen positive cells in Alzheimer’s disease brain tissue. Neurosci. Lett. 1988, 91, 259–264. [Google Scholar] [CrossRef]
- Browne, T.C.; McQuillan, K.; McManus, R.M.; O’Reilly, J.A.; Mills, K.H.; Lynch, M.A. IFN-γ Production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J. Immunol. 2013, 190, 2241–2251. [Google Scholar] [CrossRef]
- Ferretti, M.T.; Merlini, M.; Späni, C.; Gericke, C.; Schweizer, N.; Enzmann, G.; Engelhardt, B.; Kulic, L.; Suter, T.; Nitsch, R.M. T-cell brain infiltration and immature antigen-presenting cells in transgenic models of Alzheimer’s disease-like cerebral amyloidosis. Brain Behav. Immun. 2016, 54, 211–225. [Google Scholar] [CrossRef]
- Monsonego, A.; Imitola, J.; Petrovic, S.; Zota, V.; Nemirovsky, A.; Baron, R.; Fisher, Y.; Owens, T.; Weiner, H.L. Abeta-induced meningoencephalitis is IFN-gamma-dependent and is associated with T cell-dependent clearance of Abeta in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5048–5053. [Google Scholar] [CrossRef]
- Fisher, Y.; Strominger, I.; Biton, S.; Nemirovsky, A.; Baron, R.; Monsonego, A. Th1 polarization of T cells injected into the cerebrospinal fluid induces brain immunosurveillance. J. Immunol. 2014, 192, 92–102. [Google Scholar] [CrossRef]
- Cao, C.; Arendash, G.W.; Dickson, A.; Mamcarz, M.B.; Lin, X.; Ethell, D.W. Abeta-specific Th2 cells provide cognitive and pathological benefits to Alzheimer’s mice without infiltrating the CNS. Neurobiol. Dis. 2009, 34, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Bettcher, B.M.; Tansey, M.G.; Dorothée, G.; Heneka, M.T. Peripheral and central immune system crosstalk in Alzheimer disease—A research prospectus. Nat. Rev. Neurol. 2021, 17, 689–701. [Google Scholar] [CrossRef]
- Ewers, M.; Franzmeier, N.; Suárez-Calvet, M.; Morenas-Rodriguez, E.; Caballero, M.A.A.; Kleinberger, G.; Piccio, L.; Cruchaga, C.; Deming, Y.; Dichgans, M.; et al. Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci. Transl. Med. 2019, 11, eaav6221. [Google Scholar] [CrossRef] [PubMed]
- Pillai, J.A.; Bena, J.; Bebek, G.; Bekris, L.M.; Bonner-Jackson, A.; Kou, L.; Pai, A.; Sørensen, L.; Neilsen, M.; Rao, S.M.; et al. Inflammatory pathway analytes predicting rapid cognitive decline in MCI stage of Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2020, 7, 1225–1239. [Google Scholar] [CrossRef] [PubMed]
- Westin, K.; Buchhave, P.; Nielsen, H.; Minthon, L.; Janciauskiene, S.; Hansson, O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer’s disease. PLoS ONE 2012, 7, e30525. [Google Scholar] [CrossRef] [PubMed]
- Taipa, R.; das Neves, S.P.; Sousa, A.L.; Fernandes, J.; Pinto, C.; Correia, A.P.; Santos, E.; Pinto, P.S.; Carneiro, P.; Costa, P.; et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol. Aging 2019, 76, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.F.; Savard, M.; Poirier, J.; Labonté, A.; Rosa-Neto, P.; Weitz, T.M.; Town, T.; Breitner, J.; Initiative, A.S.D.N.; Group, P.-A.R. Bi-directional Association of Cerebrospinal Fluid Immune Markers with Stage of Alzheimer’s Disease Pathogenesis. J. Alzheimer’s Dis. 2018, 63, 577–590. [Google Scholar] [CrossRef]
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.P.; Rivest, S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 2006, 49, 489–502. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef]
- Naert, G.; Rivest, S. CC chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 6208–6220. [Google Scholar] [CrossRef]
- Varvel, N.H.; Grathwohl, S.A.; Degenhardt, K.; Resch, C.; Bosch, A.; Jucker, M.; Neher, J.J. Replacement of brain-resident myeloid cells does not alter cerebral amyloid-β deposition in mouse models of Alzheimer’s disease. J. Exp. Med. 2015, 212, 1803–1809. [Google Scholar] [CrossRef]
- Prokop, S.; Miller, K.R.; Drost, N.; Handrick, S.; Mathur, V.; Luo, J.; Wegner, A.; Wyss-Coray, T.; Heppner, F.L. Impact of peripheral myeloid cells on amyloid-β pathology in Alzheimer’s disease-like mice. J. Exp. Med. 2015, 212, 1811–1818. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Minogue, A.M.; Lyons, A.; Jones, R.S.; Browne, T.C.; Costello, D.A.; Denieffe, S.; O’Sullivan, C.; Connor, T.J.; Lynch, M.A. Glial Activation in AβPP/PS1 Mice is Associated with Infiltration of IFNγ-Producing Cells. J. Alzheimer’s Dis. 2013, 37, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Poduslo, J.F.; Curran, G.L.; Berg, C.T. Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc. Natl. Acad. Sci. USA 1994, 91, 5705–5709. [Google Scholar] [CrossRef]
- Bacher, M.; Depboylu, C.; Du, Y.; Noelker, C.; Oertel, W.H.; Behr, T.; Henriksen, G.; Behe, M.; Dodel, R. Peripheral and central biodistribution of (111)In-labeled anti-beta-amyloid autoantibodies in a transgenic mouse model of Alzheimer’s disease. Neurosci. Lett. 2009, 449, 240–245. [Google Scholar] [CrossRef]
- Bettcher, B.M.; Johnson, S.C.; Fitch, R.; Casaletto, K.B.; Heffernan, K.S.; Asthana, S.; Zetterberg, H.; Blennow, K.; Carlsson, C.M.; Neuhaus, J.; et al. Cerebrospinal Fluid and Plasma Levels of Inflammation Differentially Relate to CNS Markers of Alzheimer’s Disease Pathology and Neuronal Damage. J. Alzheimer’s Dis. 2018, 62, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Popp, J.; Oikonomidi, A.; Tautvydaitė, D.; Dayon, L.; Bacher, M.; Migliavacca, E.; Henry, H.; Kirkland, R.; Severin, I.; Wojcik, J.; et al. Markers of neuroinflammation associated with Alzheimer’s disease pathology in older adults. Brain Behav. Immun. 2017, 62, 203–211. [Google Scholar] [CrossRef]
- Whelan, C.D.; Mattsson, N.; Nagle, M.W.; Vijayaraghavan, S.; Hyde, C.; Janelidze, S.; Stomrud, E.; Lee, J.; Fitz, L.; Samad, T.A.; et al. Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer’s disease. Acta Neuropathol. Commun. 2019, 7, 169. [Google Scholar] [CrossRef]
- Reitz, C. Toward precision medicine in Alzheimer’s disease. Ann. Transl. Med. 2016, 4, 107. [Google Scholar] [CrossRef]
- Leoni, V. The effect of apolipoprotein E (ApoE) genotype on biomarkers of amyloidogenesis, tau pathology and neurodegeneration in Alzheimer’s disease. Clin. Chem. Lab. Med. 2011, 49, 375–383. [Google Scholar] [CrossRef]
- Cheng-Hathaway, P.J.; Reed-Geaghan, E.G.; Jay, T.R.; Casali, B.T.; Bemiller, S.M.; Puntambekar, S.S.; von Saucken, V.E.; Williams, R.Y.; Karlo, J.C.; Moutinho, M.; et al. The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 29. [Google Scholar] [CrossRef]
- Silva, M.V.F.; Loures, C.M.G.; Alves, L.C.V.; de Souza, L.C.; Borges, K.B.G.; Carvalho, M.D.G. Alzheimer’s disease: Risk factors and potentially protective measures. J. Biomed. Sci. 2019, 26, 33. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Boutajangout, A. Vaccination as a therapeutic approach to Alzheimer’s disease. Mt. Sinai J. Med. 2010, 77, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ma, Q.J. Prophylactic and therapeutic vaccines against Alzheimer’s disease. Sheng Wu Gong Cheng Xue Bao 2003, 19, 641–645. [Google Scholar]
- Vickers, J.C. A vaccine against Alzheimer’s disease: Developments to date. Drugs Aging 2002, 19, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. How plausible is an Alzheimer’s disease vaccine? Expert. Opin. Drug Discov. 2020, 15, 1–6. [Google Scholar] [CrossRef]
- Mantile, F.; Prisco, A. Vaccination against β-Amyloid as a Strategy for the Prevention of Alzheimer’s Disease. Biology 2020, 9, 425. [Google Scholar] [CrossRef]
- Furlan, R.; Brambilla, E.; Sanvito, F.; Roccatagliata, L.; Olivieri, S.; Bergami, A.; Pluchino, S.; Uccelli, A.; Comi, G.; Martino, G. Vaccination with amyloid-beta peptide induces autoimmune encephalomyelitis in C57/BL6 mice. Brain 2003, 126, 285–291. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Cacabelos, R.; Cacabelos, P.; Torrelas, C. Personalized Medicine of Alzheimer’s Disease. In Handbook of Pharmacogenomics and Stratified Medicine; Elsevier: Amsterdam, The Netherlands, 2014; Volume 27, pp. 563–615. [Google Scholar]
- Wraith, D.C. The Future of Immunotherapy: A 20-Year Perspective. Front. Immunol. 2017, 8, 1668. [Google Scholar] [CrossRef]
- Davis, I.D. An overview of cancer immunotherapy. Immunol. Cell Biol. 2000, 78, 179–195. [Google Scholar] [CrossRef]
- Brody, D.L.; Holtzman, D.M. Active and passive immunotherapy for neurodegenerative disorders. Annu. Rev. Neurosci. 2008, 31, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Oelschlaeger, T.A. Bacteria as tumor therapeutics? Bioeng. Bugs 2010, 1, 146–147. [Google Scholar] [CrossRef] [PubMed]
- Georgievska, B.; Gustavsson, S.; Lundkvist, J.; Neelissen, J.; Eketjäll, S.; Ramberg, V.; Bueters, T.; Agerman, K.; Juréus, A.; Svensson, S.; et al. Revisiting the peripheral sink hypothesis: Inhibiting BACE1 activity in the periphery does not alter β-amyloid levels in the CNS. J. Neurochem. 2015, 132, 477–486. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Inflammation, anti-inflammatory agents and Alzheimer disease: The last 12 years. J. Alzheimer’s Dis. 2006, 9, 271–276. [Google Scholar] [CrossRef] [PubMed]
- in t’ Veld, B.A.; Ruitenberg, A.; Hofman, A.; Launer, L.J.; van Duijn, C.M.; Stijnen, T.; Breteler, M.M.; Stricker, B.H. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N. Engl. J. Med. 2001, 345, 1515–1521. [Google Scholar] [CrossRef]
- Andrieu, S.; Coley, N.; Lovestone, S.; Aisen, P.S.; Vellas, B. Prevention of sporadic Alzheimer’s disease: Lessons learned from clinical trials and future directions. Lancet Neurol. 2015, 14, 926–944. [Google Scholar] [CrossRef]
- Adapt-Fs Research Group. Follow-up evaluation of cognitive function in the randomized Alzheimer’s Disease Anti-inflammatory Prevention Trial and its Follow-up Study. Alzheimer’s Dement 2015, 11, 216–225.e1. [Google Scholar] [CrossRef]
- Sudduth, T.L.; Greenstein, A.; Wilcock, D.M. Intracranial injection of Gammagard, a human IVIg, modulates the inflammatory response of the brain and lowers Aβ in APP/PS1 mice along a different time course than anti-Aβ antibodies. J. Neurosci. 2013, 33, 9684–9692. [Google Scholar] [CrossRef]
- Counts, S.E.; Ray, B.; Mufson, E.J.; Perez, S.E.; He, B.; Lahiri, D.K. Intravenous immunoglobulin (IVIG) treatment exerts antioxidant and neuropreservatory effects in preclinical models of Alzheimer’s disease. J. Clin. Immunol. 2014, 34 (Suppl. 1), S80–S85. [Google Scholar] [CrossRef]
- Loeffler, D.A. Intravenous immunoglobulin and Alzheimer’s disease: What now? J. Neuroinflamm. 2013, 10, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodel, R.; Rominger, A.; Bartenstein, P.; Barkhof, F.; Blennow, K.; Förster, S.; Winter, Y.; Bach, J.P.; Popp, J.; Alferink, J.; et al. Intravenous immunoglobulin for treatment of mild-to-moderate Alzheimer’s disease: A phase 2, randomised, double-blind, placebo-controlled, dose-finding trial. Lancet Neurol 2013, 12, 233–243. [Google Scholar] [CrossRef]
- Relkin, N. Clinical trials of intravenous immunoglobulin for Alzheimer’s disease. J. Clin. Immunol. 2014, 34 (Suppl. 1), S74–S79. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, K.P.; Sompol, P.; Kannarkat, G.T.; Chang, J.; Sniffen, L.; Wildner, M.E.; Norris, C.M.; Tansey, M.G. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 2017, 102, 81–95. [Google Scholar] [CrossRef]
- Butchart, J.; Brook, L.; Hopkins, V.; Teeling, J.; Püntener, U.; Culliford, D.; Sharples, R.; Sharif, S.; McFarlane, B.; Raybould, R.; et al. Etanercept in Alzheimer disease: A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 2015, 84, 2161–2168. [Google Scholar] [CrossRef]
- Sha, S.J.; Deutsch, G.K.; Tian, L.; Richardson, K.; Coburn, M.; Gaudioso, J.L.; Marcal, T.; Solomon, E.; Boumis, A.; Bet, A.; et al. Safety, Tolerability, and Feasibility of Young Plasma Infusion in the Plasma for Alzheimer Symptom Amelioration Study: A Randomized Clinical Trial. JAMA Neurol. 2019, 76, 35–40. [Google Scholar] [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef]
- Winblad, B.; Graf, A.; Riviere, M.E.; Andreasen, N.; Ryan, J.M. Active immunotherapy options for Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 7. [Google Scholar] [CrossRef]
- Kensil, C.R.; Patel, U.; Lennick, M.; Marciani, D. Separation and characterization of saponins with adjuvant activity from Quillaja saponaria Molina cortex. J. Immunol. 1991, 146, 431–437. [Google Scholar]
- Fox, N.C.; Black, R.S.; Gilman, S.; Rossor, M.N.; Griffith, S.G.; Jenkins, L.; Koller, M. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 2005, 64, 1563–1572. [Google Scholar] [CrossRef]
- Vandenberghe, R.; Riviere, M.E.; Caputo, A.; Sovago, J.; Maguire, R.P.; Farlow, M.; Marotta, G.; Sanchez-Valle, R.; Scheltens, P.; Ryan, J.M.; et al. Active Aβ immunotherapy CAD106 in Alzheimer’s disease: A phase 2b study. Alzheimer’s Dement 2017, 3, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Winblad, B.; Andreasen, N.; Minthon, L.; Floesser, A.; Imbert, G.; Dumortier, T.; Maguire, R.P.; Blennow, K.; Lundmark, J.; Staufenbiel, M.; et al. Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer’s disease: Randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012, 11, 597–604. [Google Scholar] [CrossRef]
- Lacosta, A.M.; Pascual-Lucas, M.; Pesini, P.; Casabona, D.; Pérez-Grijalba, V.; Marcos-Campos, I.; Sarasa, L.; Canudas, J.; Badi, H.; Monleón, I.; et al. Safety, tolerability and immunogenicity of an active anti-Aβ. Alzheimer’s Res. Ther. 2018, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- NIH. Safety and Immunogenicity of Repeated Doses of ABvac40 in Patients with a-MCI or Vm-AD. Available online: https://clinicaltrials.gov/ct2/show/record/NCT03461276 (accessed on 17 September 2021).
- Muhs, A.; Hickman, D.T.; Pihlgren, M.; Chuard, N.; Giriens, V.; Meerschman, C.; van der Auwera, I.; van Leuven, F.; Sugawara, M.; Weingertner, M.C.; et al. Liposomal vaccines with conformation-specific amyloid peptide antigens define immune response and efficacy in APP transgenic mice. Proc. Natl. Acad. Sci. USA 2007, 104, 9810–9815. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.B.; Montufar, N.; DeLeon, J.; Pinkhasov, A.; Gomolin, I.H.; Glass, A.D.; Arain, H.A.; Stecker, M.M. Alzheimer Disease Clinical Trials Targeting Amyloid: Lessons Learned From Success in Mice and Failure in Humans. Neurologist 2021, 26, 52–61. [Google Scholar] [CrossRef]
- Wang, C.Y.; Wang, P.N.; Chiu, M.J.; Finstad, C.L.; Lin, F.; Lynn, S.; Tai, Y.H.; De Fang, X.; Zhao, K.; Hung, C.H.; et al. UB-311, a novel UBITh. Alzheimer’s Dement 2017, 3, 262–272. [Google Scholar] [CrossRef]
- Bigaeva, E.; Doorn, E.; Liu, H.; Hak, E. Meta-Analysis on Randomized Controlled Trials of Vaccines with QS-21 or ISCOMATRIX Adjuvant: Safety and Tolerability. PLoS ONE 2016, 11, e0154757. [Google Scholar] [CrossRef]
- Ketter, N.; Liu, E.; Di, J.; Honig, L.S.; Lu, M.; Novak, G.; Werth, J.; LePrince Leterme, G.; Shadman, A.; Brashear, H.R. A Randomized, Double-Blind, Phase 2 Study of the Effects of the Vaccine Vanutide Cridificar with QS-21 Adjuvant on Immunogenicity, Safety and Amyloid Imaging in Patients with Mild to Moderate Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2016, 3, 192–201. [Google Scholar] [CrossRef]
- Davtyan, H.; Ghochikyan, A.; Petrushina, I.; Hovakimyan, A.; Davtyan, A.; Poghosyan, A.; Marleau, A.M.; Movsesyan, N.; Kiyatkin, A.; Rasool, S.; et al. Immunogenicity, efficacy, safety, and mechanism of action of epitope vaccine (Lu AF20513) for Alzheimer’s disease: Prelude to a clinical trial. J. Neurosci. 2013, 33, 4923–4934. [Google Scholar] [CrossRef]
- NIH. Safety and Immunogenicity of Repeated Doses of ABvac40 in Patients with a-MCI or Vm-AD. Available online: https://clinicaltrials.gov/ct2/show/NCT03461276 (accessed on 17 September 2021).
- NIH. A Study of ACI-24 in Adults with Down Syndrome. Available online: https://clinicaltrials.gov/ct2/show/NCT04373616 (accessed on 17 September 2021).
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimer’s Dement. 2019, 5, 272–293. [Google Scholar] [CrossRef]
- NIH. Evaluate the Safety, Tolerability, Immunogenicity and Efficacy of UB-311 in Mild Alzheimer’s Disease (AD) Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT02551809 (accessed on 17 September 2021).
- NIH. A Study of V950 in People with Alzheimer Disease (V950-001 AM7). Available online: https://clinicaltrials.gov/ct2/show/NCT00464334 (accessed on 17 September 2021).
- Hull, M.; Sadowsky, C.; Arai, H.; Le Prince Leterme, G.; Holstein, A.; Booth, K.; Peng, Y.; Yoshiyama, T.; Suzuki, H.; Ketter, N.; et al. Long-Term Extensions of Randomized Vaccination Trials of ACC-001 and QS-21 in Mild to Moderate Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, F.; Sadowsky, C.; Holstein, A.; Leterme, G.L.P.; Peng, Y.; Jackson, N.; Fox, N.C.; Ketter, N.; Liu, E.; Ryan, J.M.; et al. Two Phase 2 Multiple Ascending-Dose Studies of Vanutide Cridificar (ACC-001) and QS-21 Adjuvant in Mild-to-Moderate Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 51, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- NIH. Study with Lu AF20513 in Patients with Mild Alzheimer’s Disease (AD) or Mild Cognitive Impairment (MCI) Due to AD. Available online: https://clinicaltrials.gov/ct2/show/NCT03819699 (accessed on 17 September 2021).
- Spencer, B.; Masliah, E. Immunotherapy for Alzheimer’s disease: Past, present and future. Front. Aging Neurosci. 2014, 6, 114. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.S.; Cashman, N.R. Passive immunotherapies targeting Aβ and tau in Alzheimer’s disease. Neurobiol. Dis. 2020, 144, 105010. [Google Scholar] [CrossRef]
- Delnomdedieu, M.; Duvvuri, S.; Li, D.J.; Atassi, N.; Lu, M.; Brashear, H.R.; Liu, E.; Ness, S.; Kupiec, J.W. First-In-Human safety and long-term exposure data for AAB-003 (PF-05236812) and biomarkers after intravenous infusions of escalating doses in patients with mild to moderate Alzheimer’s disease. Alzheimer’s Res. Ther. 2016, 8, 12. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Gilman, S.; Fox, N.C.; Blennow, K.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Doody, R.; van Dyck, C.H.; et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 2009, 73, 2061–2070. [Google Scholar] [CrossRef]
- Bohrmann, B.; Baumann, K.; Benz, J.; Gerber, F.; Huber, W.; Knoflach, F.; Messer, J.; Oroszlan, K.; Rauchenberger, R.; Richter, W.F.; et al. Gantenerumab: A novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J. Alzheimer’s Dis. 2012, 28, 49–69. [Google Scholar] [CrossRef]
- Piazza, F.; Winblad, B. Amyloid-Related Imaging Abnormalities (ARIA) in Immunotherapy Trials for Alzheimer’s Disease: Need for Prognostic Biomarkers? J. Alzheimer’s Dis. 2016, 52, 417–420. [Google Scholar] [CrossRef]
- Yang, T.; Dang, Y.; Ostaszewski, B.; Mengel, D.; Steffen, V.; Rabe, C.; Bittner, T.; Walsh, D.M.; Selkoe, D.J. Target engagement in an alzheimer trial: Crenezumab lowers amyloid β oligomers in cerebrospinal fluid. Ann. Neurol. 2019, 86, 215–224. [Google Scholar] [CrossRef]
- Yoshida, K.; Moein, A.; Bittner, T.; Ostrowitzki, S.; Lin, H.; Honigberg, L.; Jin, J.Y.; Quartino, A. Pharmacokinetics and pharmacodynamic effect of crenezumab on plasma and cerebrospinal fluid beta-amyloid in patients with mild-to-moderate Alzheimer’s disease. Alzheimer’s Res. Ther. 2020, 12, 16. [Google Scholar] [CrossRef]
- Guthrie, H.; Honig, L.S.; Lin, H.; Sink, K.M.; Blondeau, K.; Quartino, A.; Dolton, M.; Carrasco-Triguero, M.; Lian, Q.; Bittner, T.; et al. Safety, Tolerability, and Pharmacokinetics of Crenezumab in Patients with Mild-to-Moderate Alzheimer’s Disease Treated with Escalating Doses for up to 133 Weeks. J. Alzheimer’s Dis. 2020, 76, 967–979. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Cohen, S.; van Dyck, C.H.; Brody, M.; Curtis, C.; Cho, W.; Ward, M.; Friesenhahn, M.; Rabe, C.; Brunstein, F.; et al. ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 2018, 90, e1889–e1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salloway, S.; Honigberg, L.A.; Cho, W.; Ward, M.; Friesenhahn, M.; Brunstein, F.; Quartino, A.; Clayton, D.; Mortensen, D.; Bittner, T.; et al. Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti-amyloid-beta antibody double-blind, placebo-controlled, randomized phase II study in mild-to-moderate Alzheimer’s disease (BLAZE). Alzheimer’s Res. Ther. 2018, 10, 96. [Google Scholar] [CrossRef] [PubMed]
- Sumner, I.L.; Edwards, R.A.; Asuni, A.A.; Teeling, J.L. Antibody Engineering for Optimized Immunotherapy in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 254. [Google Scholar] [CrossRef]
- Farlow, M.; Arnold, S.E.; van Dyck, C.H.; Aisen, P.S.; Snider, B.J.; Porsteinsson, A.P.; Friedrich, S.; Dean, R.A.; Gonzales, C.; Sethuraman, G.; et al. Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimer’s Dement 2012, 8, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Siemers, E.R.; Sundell, K.L.; Carlson, C.; Case, M.; Sethuraman, G.; Liu-Seifert, H.; Dowsett, S.A.; Pontecorvo, M.J.; Dean, R.A.; Demattos, R. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimer’s Dement 2016, 12, 110–120. [Google Scholar] [CrossRef]
- Dhillon, S. Aducanumab: First Approval. Drugs 2021, 81, 1437–1443. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to watch in 2020. MAbs 2020, 12, 1703531. [Google Scholar] [CrossRef]
- Schneider, L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol. 2020, 19, 111–112. [Google Scholar] [CrossRef]
- Mullard, A. Controversial Alzheimer’s drug approval could affect other diseases. Nature 2021, 595, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Logovinsky, V.; Satlin, A.; Lai, R.; Swanson, C.; Kaplow, J.; Osswald, G.; Basun, H.; Lannfelt, L. Safety and tolerability of BAN2401--a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimer’s Res. Ther. 2016, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- NIH. A Study to Confirm Safety and Efficacy of Lecanemab in Participants with Early Alzheimer’s Disease (Clarity AD). Available online: https://clinicaltrials.gov/ct2/show/NCT03887455 (accessed on 18 September 2021).
- Abushouk, A.I.; Elmaraezy, A.; Aglan, A.; Salama, R.; Fouda, S.; Fouda, R.; AlSafadi, A.M. Bapineuzumab for mild to moderate Alzheimer’s disease: A meta-analysis of randomized controlled trials. BMC Neurol. 2017, 17, 66. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Frisardi, V.; Imbimbo, B.P.; D’Onofrio, G.; Pietrarossa, G.; Seripa, D.; Pilotto, A.; Solfrizzi, V. Bapineuzumab: Anti-β-amyloid monoclonal antibodies for the treatment of Alzheimer’s disease. Immunotherapy 2010, 2, 767–782. [Google Scholar] [CrossRef]
- NIH. Safety and Efficacy Study of Gantenerumab in Participants with Early Alzheimer’s Disease (AD). Available online: https://clinicaltrials.gov/ct2/show/NCT03443973 (accessed on 18 September 2021).
- Doggrell, S.A. Grasping at straws: The failure of solanezumab to modify mild Alzheimer’s disease. Expert. Opin. Biol. Ther. 2018, 18, 1189–1192. [Google Scholar] [CrossRef]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer disease and aducanumab: Adjusting our approach. Nat. Rev. Neurol. 2019, 15, 365–366. [Google Scholar] [CrossRef]
- Maciejko, L.; Smalley, M.; Goldman, A. Cancer Immunotherapy and Personalized Medicine: Emerging Technologies and Biomarker-Based Approaches. J. Mol. Biomark. Diagn. 2017, 8, 350. [Google Scholar] [CrossRef]
- Tyson, R.J.; Park, C.C.; Powell, J.R.; Patterson, J.H.; Weiner, D.; Watkins, P.B.; Gonzalez, D. Precision Dosing Priority Criteria: Drug, Disease, and Patient Population Variables. Front. Pharmacol. 2020, 11, 420. [Google Scholar] [CrossRef]
- Adamaki, M.; Zoumpourlis, V. Immunotherapy as a Precision Medicine Tool for the Treatment of Prostate Cancer. Cancers 2021, 13, 173. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.; Salmon, D.; Galasko, D.; Masliah, E.; Katzman, R.; DeTeresa, R.; Thal, L.; Pay, M.M.; Hofstetter, R.; Klauber, M. The Lewy body variant of Alzheimer’s disease: A clinical and pathologic entity. Neurology 1990, 40, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Román, G.C.; Rogers, S.J. Donepezil: A clinical review of current and emerging indications. Expert. Opin. Pharmacother. 2004, 5, 161–180. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, C.; Jiménez Caballero, P.E.; Alfonso, V.; González-Adalid, M. Current treatments of Alzheimer disease: Are main caregivers satisfied with the drug treatments received by their patients? Dement Geriatr. Cogn. Disord. 2009, 28, 196–205. [Google Scholar] [CrossRef]
- Mathur, S.; Sutton, J. Personalized medicine could transform healthcare. Biomed. Rep. 2017, 7, 3–5. [Google Scholar] [CrossRef]
- Barman, N.C.; Khan, N.M.; Islam, M.; Nain, Z.; Roy, R.K.; Haque, A.; Barman, S.K. CRISPR-Cas9: A Promising Genome Editing Therapeutic Tool for Alzheimer’s Disease-A Narrative Review. Neurol. Ther. 2020, 9, 419–434. [Google Scholar] [CrossRef]
- Ivanisevic, J.; Thomas, A. Metabolomics as a Tool to Understand Pathophysiological Processes. Methods Mol. Biol. 2018, 1730, 3–28. [Google Scholar] [CrossRef]
- Reiman, E.M.; Langbaum, J.B.; Fleisher, A.S.; Caselli, R.J.; Chen, K.; Ayutyanont, N.; Quiroz, Y.T.; Kosik, K.S.; Lopera, F.; Tariot, P.N. Alzheimer’s Prevention Initiative: A plan to accelerate the evaluation of presymptomatic treatments. J. Alzheimer’s Dis. 2011, 26 (Suppl. 3), 321–329. [Google Scholar] [CrossRef]
- Mills, S.M.; Mallmann, J.; Santacruz, A.M.; Fuqua, A.; Carril, M.; Aisen, P.S.; Althage, M.C.; Belyew, S.; Benzinger, T.L.; Brooks, W.S.; et al. Preclinical trials in autosomal dominant AD: Implementation of the DIAN-TU trial. Rev. Neurol. 2013, 169, 737–743. [Google Scholar] [CrossRef]
- Sperling, R.A.; Rentz, D.M.; Johnson, K.A.; Karlawish, J.; Donohue, M.; Salmon, D.P.; Aisen, P. The A4 study: Stopping AD before symptoms begin? Sci. Transl. Med. 2014, 6, 228fs213. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zieneldien, T.; Kim, J.; Sawmiller, D.; Cao, C. The Immune System as a Therapeutic Target for Alzheimer’s Disease. Life 2022, 12, 1440. https://doi.org/10.3390/life12091440
Zieneldien T, Kim J, Sawmiller D, Cao C. The Immune System as a Therapeutic Target for Alzheimer’s Disease. Life. 2022; 12(9):1440. https://doi.org/10.3390/life12091440
Chicago/Turabian StyleZieneldien, Tarek, Janice Kim, Darrell Sawmiller, and Chuanhai Cao. 2022. "The Immune System as a Therapeutic Target for Alzheimer’s Disease" Life 12, no. 9: 1440. https://doi.org/10.3390/life12091440
APA StyleZieneldien, T., Kim, J., Sawmiller, D., & Cao, C. (2022). The Immune System as a Therapeutic Target for Alzheimer’s Disease. Life, 12(9), 1440. https://doi.org/10.3390/life12091440