
Detection of Recombinant Proteins SOX2 and OCT4 Interacting in HEK293T Cells Using Real-Time Quantitative PCR


 and
and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
- verify the presence or absence of the expression of the endogenous proteins SOX2 and OCT4;
- test the specificity of the BirA/BAP pair in the presence of the endogenous background biotinylation;
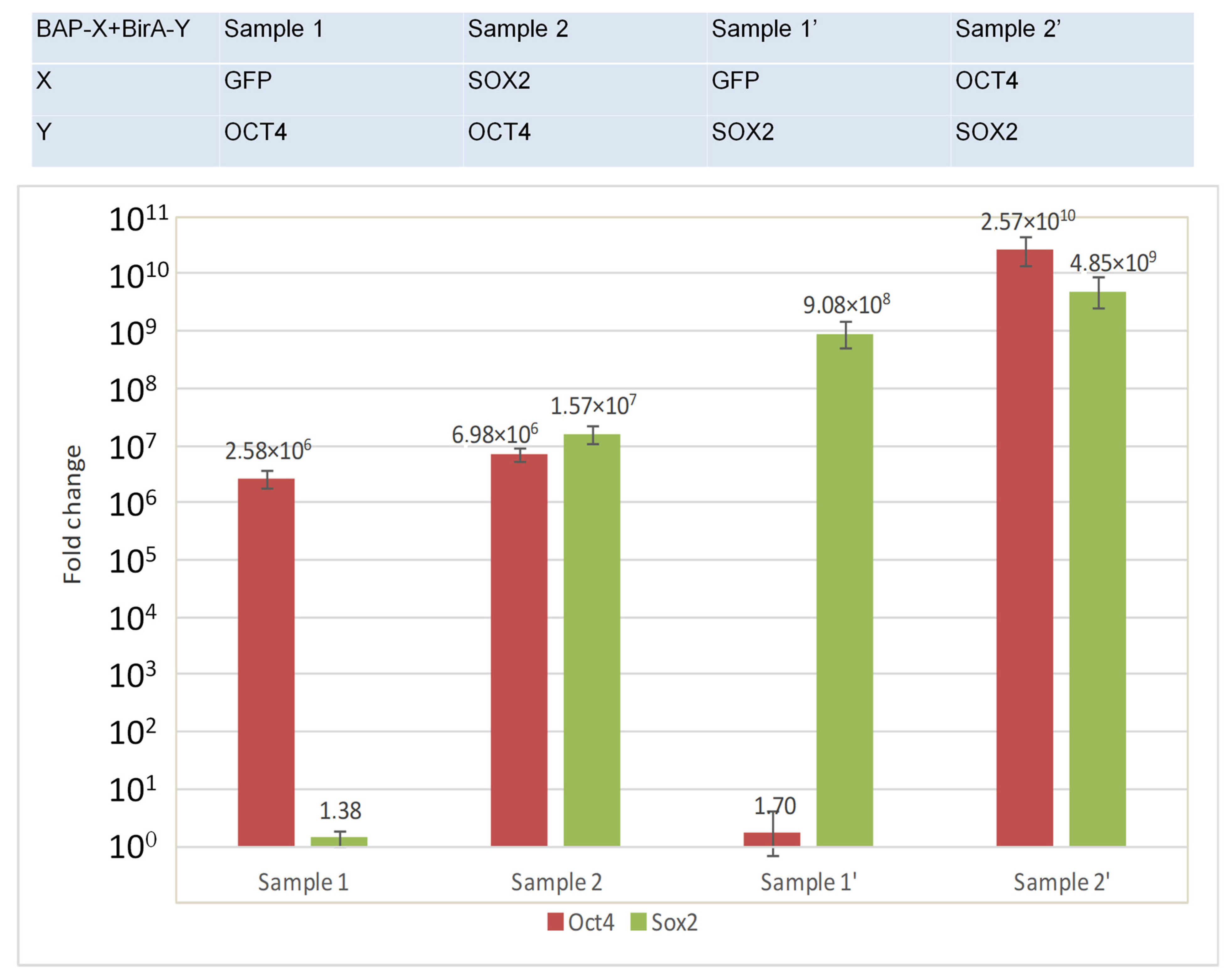
- compare the amount of BirA-Y (SOX2, OCT4) in the experiment (interacting proteins) and in the control (non-interacting proteins) by real-time qRT-PCR.
2. Materials and Methods
2.1. Materials
2.2. Primers Used for Real-Time qRT-PCR
2.3. Cell Culture
2.4. Transient Transfection by LIPOFECTAMINE 2000 (General Procedure, X,Y = SOX2, OCT4)
2.5. Biotin Labeling and Cell Harvesting
2.6. Western Blotting (General Procedure, X,Y = SOX2, OCT4)
2.7. RNA Isolation with TRIZOL Reagent from HEK293T
2.8. Reverse Transcription (General Procedure, X,Y = SOX2, OCT4)
2.9. Real-Time qRT-PCR
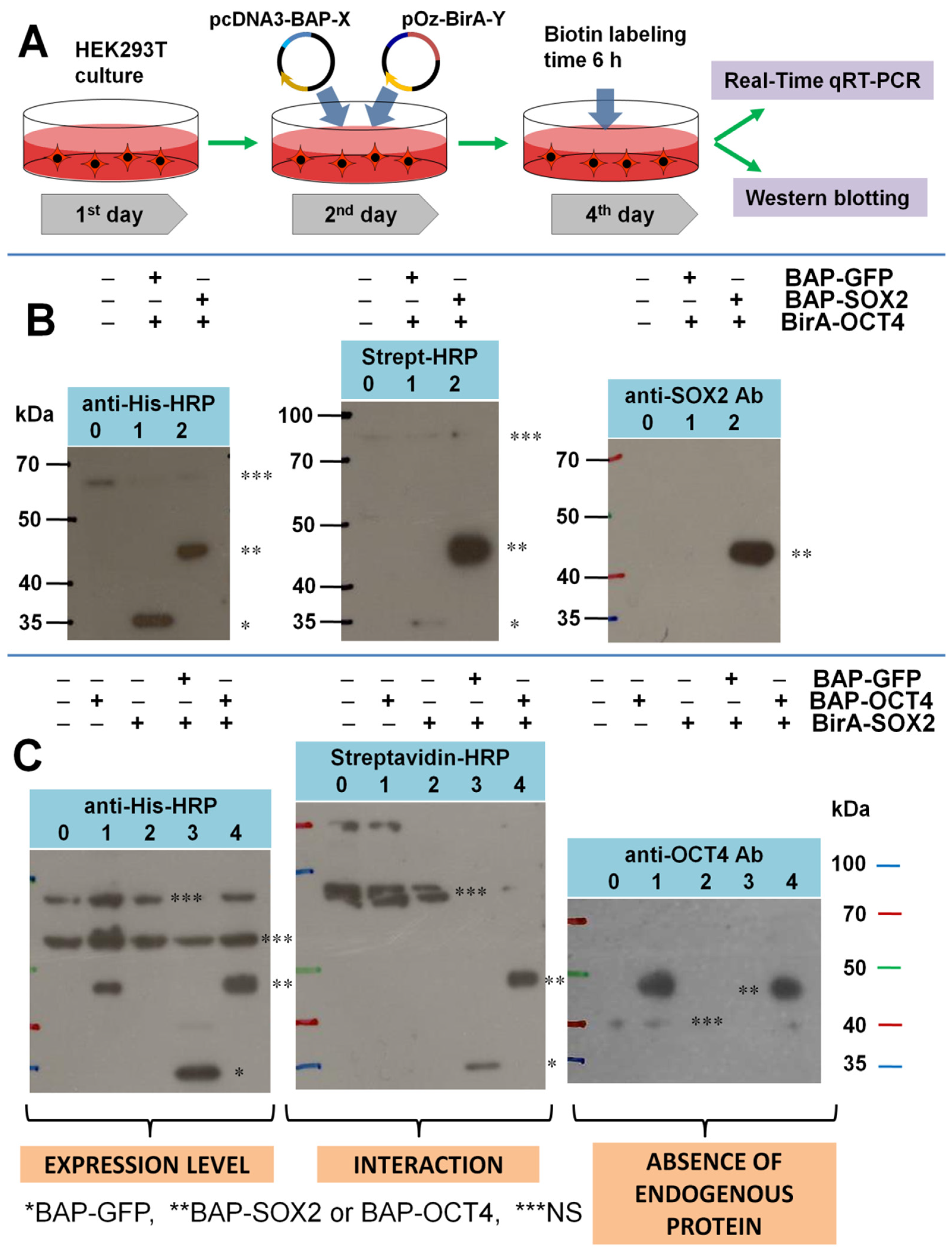
3. Results and Discussion
- Testing the level of expression of endogenous SOX2 and OCT4 proteins in cells;
- Introducing additional controls showing the absence of BAP biotinylation due to endogenous biotin ligases from HEK293T cells;
- Quantitatively comparing the expression level of BirA fusions in the control and experiment using real-time qRT-PCR.
3.1. Testing the Level of Expression of Endogenous SOX2 and OCT4 Proteins in Cells
3.2. Testing the Specificity of Biotinylation for the BAP/BirA Pair in HEK293T Cells
3.3. Quantitative Comparison of the Expression Level of BirA-Y (Y = SOX2, OCT4) Fusions Using Real-Time qRT-PCR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marcilla, M.; Albar, J.P. Quantitative proteomics: A strategic ally to map protein interaction networks. IUBMB Life 2013, 65, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Smits, A.H.; Vermeulen, M. Characterizing Protein-Protein Interactions Using Mass Spectrometry: Challenges and Opportunities. Trends Biotechnol. 2016, 34, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Kido, K.; Yamanaka, S.; Nakano, S.; Motani, K.; Shinohara, S.; Nozawa, A.; Kosako, H.; Ito, S.; Sawasaki, T. AirID, a novel proximity biotinylation enzyme, for analysis of protein-protein interactions. Elife 2020, 9, e54983. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, M.; Majzoub, K.; Rao, D.S.; Neela, P.H.; Zarnegar, B.J.; Mondal, S.; Roth, J.G.; Gai, H.; Kovalski, J.R.; Siprashvili, Z.; et al. RNA-protein interaction detection in living cells. Nat. Methods 2018, 15, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Villasenor, R.; Pfaendler, R.; Ambrosi, C.; Butz, S.; Giuliani, S.; Bryan, E.; Sheahan, T.W.; Gable, A.L.; Schmolka, N.; Manzo, M.; et al. ChromID identifies the protein interactome at chromatin marks. Nat. Biotechnol. 2020, 38, 728–736. [Google Scholar] [CrossRef]
- Go, C.D.; Knight, J.D.R.; Rajasekharan, A.; Rathod, B.; Hesketh, G.G.; Abe, K.T.; Youn, J.Y.; Samavarchi-Tehrani, P.; Zhang, H.; Zhu, L.Y.; et al. A proximity-dependent biotinylation map of a human cell. Nature 2021, 595, 120–124. [Google Scholar] [CrossRef]
- Samavarchi-Tehrani, P.; Samson, R.; Gingras, A.C. Proximity Dependent Biotinylation: Key Enzymes and Adaptation to Proteomics Approaches. Mol. Cell. Proteom. 2020, 19, 757–773. [Google Scholar] [CrossRef] [Green Version]
- Fairhead, M.; Howarth, M. Site-specific biotinylation of purified proteins using BirA. Methods Mol. Biol. 2015, 1266, 171–184. [Google Scholar] [CrossRef]
- Beckett, D.; Kovaleva, E.; Schatz, P.J. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999, 8, 921–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Suarez, M.; Chen, T.S.; Ting, A.Y. Protein-protein interaction detection in vitro and in cells by proximity biotinylation. J. Am. Chem. Soc. 2008, 130, 9251–9253. [Google Scholar] [CrossRef] [Green Version]
- Kulyyassov, A.; Shoaib, M.; Pichugin, A.; Kannouche, P.; Ramanculov, E.; Lipinski, M.; Ogryzko, V. PUB-MS: A Mass Spectrometry-based Method to Monitor Protein-Protein Proximity in vivo. J. Proteome Res. 2011, 10, 4416–4427. [Google Scholar] [CrossRef]
- Kulyyassov, A.; Ogryzko, V. In Vivo Quantitative Estimation of DNA-Dependent Interaction of Sox2 and Oct4 Using BirA-Catalyzed Site-Specific Biotinylation. Biomolecules 2020, 10, 142. [Google Scholar] [CrossRef] [Green Version]
- Shoaib, M.; Kulyyassov, A.; Robin, C.; Winczura, K.; Tarlykov, P.; Despas, E.; Kannouche, P.; Ramanculov, E.; Lipinski, M.; Ogryzko, V. PUB-NChIP-”in vivo biotinylation” approach to study chromatin in proximity to a protein of interest. Genome Res. 2013, 23, 331–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulyyassov, A.; Ramankulov, Y.; Ogryzko, V. Generation of Peptides for Highly Efficient Proximity Utilizing Site-Specific Biotinylation in Cells. Life 2022, 12, 300. [Google Scholar] [CrossRef]
- Jurisic, A.; Robin, C.; Tarlykov, P.; Siggens, L.; Schoell, B.; Jauch, A.; Ekwall, K.; Sorensen, C.S.; Lipinski, M.; Shoaib, M.; et al. Topokaryotyping demonstrates single cell variability and stress dependent variations in nuclear envelope associated domains. Nucleic. Acids Res. 2018, 46, e135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar, T.M.; Oksuz, O.; Saldana-Meyer, R.; Descostes, N.; Bonasio, R.; Reinberg, D. Active and Repressed Chromatin Domains Exhibit Distinct Nucleosome Segregation during DNA Replication. Cell 2019, 179, 953–963.e911. [Google Scholar] [CrossRef]
- Leon-Del-Rio, A.; Leclerc, D.; Akerman, B.; Wakamatsu, N.; Gravel, R.A. Isolation of a cDNA encoding human holocarboxylase synthetase by functional complementation of a biotin auxotroph of Escherichia coli. Proc. Natl. Acad. Sci. USA 1995, 92, 4626–4630. [Google Scholar] [CrossRef] [Green Version]
- Ingaramo, M.; Beckett, D. Selectivity in post-translational biotin addition to five human carboxylases. J. Biol. Chem. 2012, 287, 1813–1822. [Google Scholar] [CrossRef]
- Sekenova, A.E.; Baltabekova, M.Z.; Khassenov, B.B.; Ogay, V.B. Production and analyzing of polyclonal antibodies to the Sox2 recombinant transcription factor. Eurasian J. Appl. Biotechnol. 2013, 4, 16–19. [Google Scholar]
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5439. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.; Lee, D.; Schulman, A.H. FastPCR software for PCR, in silico PCR, and oligonucleotide assembly and analysis. Methods Mol. Biol. 2014, 1116, 271–302. [Google Scholar] [CrossRef]
- Kalendar, R.; Khassenov, B.; Ramankulov, Y.; Samuilova, O.; Ivanov, K.I. FastPCR: An in silico tool for fast primer and probe design and advanced sequence analysis. Genomics 2017, 109, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, B. Words of advice: Teaching enzyme kinetics. FEBS J. 2021, 288, 2068–2083. [Google Scholar] [CrossRef]
- Mechold, U.; Gilbert, C.; Ogryzko, V. Codon optimization of the BirA enzyme gene leads to higher expression and an improved efficiency of biotinylation of target proteins in mammalian cells. J. Biotechnol. 2005, 116, 245–249. [Google Scholar] [CrossRef]
- Lee, C.K.; Cheong, H.K.; Ryu, K.S.; Lee, J.I.; Lee, W.; Jeon, Y.H.; Cheong, C. Biotinoyl domain of human acetyl-CoA carboxylase: Structural insights into the carboxyl transfer mechanism. Proteins 2008, 72, 613–624. [Google Scholar] [CrossRef]
- Xiang, S.; Tong, L. Crystal structures of human and Staphylococcus aureus pyruvate carboxylase and molecular insights into the carboxyltransfer reaction. Nat. Struct. Mol. Biol. 2008, 15, 295–302. [Google Scholar] [CrossRef]
- Chu, C.H.; Cheng, D. Expression, purification, characterization of human 3-methylcrotonyl-CoA carboxylase (MCCC). Protein Expr. Purif. 2007, 53, 421–427. [Google Scholar] [CrossRef]
- Healy, S.; McDonald, M.K.; Wu, X.; Yue, W.W.; Kochan, G.; Oppermann, U.; Gravel, R.A. Structural impact of human and Escherichia coli biotin carboxyl carrier proteins on biotin attachment. Biochemistry 2010, 49, 4687–4694. [Google Scholar] [CrossRef]
- Ghosh, R.; Gilda, J.E.; Gomes, A.V. The necessity of and strategies for improving confidence in the accuracy of western blots. Expert Rev. Proteom. 2014, 11, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.S.; Lee, N.J.; Woo, S.H.; Kim, J.M.; Kim, H.M.; Jo, H.J.; Park, Y.E.; Han, M.G. Validation of real-time RT-PCR for detection of SARS-CoV-2 in the early stages of the COVID-19 outbreak in the Republic of Korea. Sci. Rep. 2021, 11, 14817. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanayev, D.; Abilmazhenova, D.; Akhmetollayev, I.; Sekenova, A.; Ogay, V.; Kulyyassov, A. Detection of Recombinant Proteins SOX2 and OCT4 Interacting in HEK293T Cells Using Real-Time Quantitative PCR. Life 2023, 13, 107. https://doi.org/10.3390/life13010107
Kanayev D, Abilmazhenova D, Akhmetollayev I, Sekenova A, Ogay V, Kulyyassov A. Detection of Recombinant Proteins SOX2 and OCT4 Interacting in HEK293T Cells Using Real-Time Quantitative PCR. Life. 2023; 13(1):107. https://doi.org/10.3390/life13010107
Chicago/Turabian StyleKanayev, Darkhan, Diana Abilmazhenova, Ilyas Akhmetollayev, Aliya Sekenova, Vyacheslav Ogay, and Arman Kulyyassov. 2023. "Detection of Recombinant Proteins SOX2 and OCT4 Interacting in HEK293T Cells Using Real-Time Quantitative PCR" Life 13, no. 1: 107. https://doi.org/10.3390/life13010107
APA StyleKanayev, D., Abilmazhenova, D., Akhmetollayev, I., Sekenova, A., Ogay, V., & Kulyyassov, A. (2023). Detection of Recombinant Proteins SOX2 and OCT4 Interacting in HEK293T Cells Using Real-Time Quantitative PCR. Life, 13(1), 107. https://doi.org/10.3390/life13010107