

Sample Size Calculation in Genetic Association Studies: A Practical Approach


Abstract
:1. Introduction
2. Sample Size Definition and Parameters for Its Calculation
2.1. Alpha and Beta Errors
- -
- Alpha (α) or the probability of Type I error occurs when H0, although actually true in the source population, is falsely rejected based on sample results (false positive result) [14]. The magnitude of this error is chosen in advance and conventionally fixed at 0.05 or 5%.
- -
- Beta (β) or the probability of Type II error occurs when H0, although false in the source population, is incorrectly not rejected according to sample results (false negative result) [14]. It is usually set at 0.20 or 20%.
2.2. Outcome
2.3. Effect Size
2.4. Minor Allele Frequency
2.5. Linkage Disequilibrium
2.6. Genetic Inheritance Model
2.7. Errors
3. Examples of the Sample Size Calculation
3.1. Example 1
Criticisms
3.2. Example 2
Criticisms
3.3. Example 3
Criticisms
4. Sample Size Calculation: A Practical Approach
4.1. Outcome/Design (Figure 1)
4.2. Hypothesis (Figure 2)
4.3. Gene G (Figure 3)
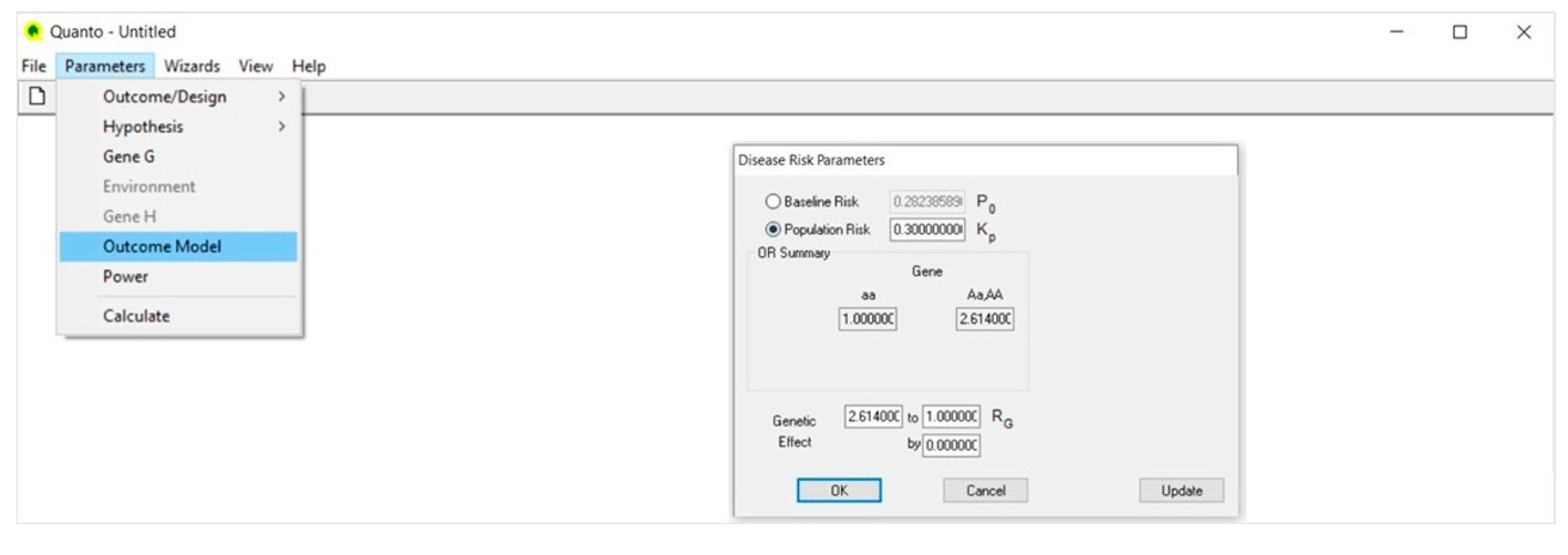
4.4. Outcome Model (Figure 4)
4.5. Power (Figure 5)
4.6. Calculate (Figure 6)
5. Software Programs for the Sample Size Calculation
- -
- QUANTO. It is a program that allows one to address hypotheses related to gene and environment only; gene–environment and gene–gene interactions. It is applicable to discrete and continuous phenotypes and deals with a large range of study designs including matched and unmatched case-control studies, case-sibling, case-parent trio and case only. In addition to the basic parameters, it requires specific parameters such as the MAF, the disease prevalence, the genetic effect size and the inheritance model [40].
- -
- ESPRESSO.G. It is an R script sample size calculator aimed at investigating the role of genes and environment on discrete and continuous phenotypes. It supports calculations for stand-alone case-control studies and for case-control analyses nested in cohort studies. In particular, this tool takes into account genotype/phenotype misclassification errors [43].
- -
- GENPWR. It is an open-source R package that defines the sample size for dichotomous or continuous outcomes in gene only and gene–environment interaction studies. By comparing the various genetic models (additive, dominant and recessive) to find the one that best fits the data, this tool allows one to calculate sample size in the presence of genetic model misspecification [44].
- -
- OSSE (Online Sample Size Estimator). It is the simplest online sample size estimator for case-control study design. The unique parameters required are the alpha error, the study power, the MAF for cases and controls and the ratio of cases to controls. No assumptions about the nature of the phenotype, the genetic inheritance model or the effect size are needed. This software does not address gene–environment interactions [16].
- -
- PAWE (Power for Association With Error). It is a sample size calculation web-based tool available on the Rockfeller site and designed for genetic case-control studies that evaluate the association between SNPs and complex discrete traits. Among the specific parameters, it requires the effect size, the MAF and the LD and allows for misclassification errors [45].
- -
- GPC (Genetic Power Calculator). It is a user-friendly computer software that performs power calculations for genetic association studies. It computes statistical power under varying disease allele frequency, disease prevalence, genotype relative risk and inheritance models. This tool requires a priori the number of cases and controls and graphically plots the relation between sample size and study power [46].
- -
- PGA (Power for Genetic Association analyses). It is a package tailored to estimate sample size in case-control studies of SNPs in unrelated individuals. However, being able to account for statistical multiple comparisons, it can also be used for fine-mapping association studies and whole-genome scans. The program provides a computational and graphical interface for the relation between statistical power and sample size under different genetic inheritance models, disease allele frequency, disease prevalence and relative risk and accounts for LD between the marker and the disease allele [47].
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Not Communicable-Diseases 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/noncommunicable-diseases (accessed on 16 September 2022).
- Khoury, M.J.; Little, J.; Gwinn, M.; Ioannidis, J.P. On the synthesis and interpretation of consistent but weak gene-disease associations in the era of genome-wide association studies. Int. J. Epidemiol. 2007, 36, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Lunetta, K.L. Genetic association studies. Circulation 2008, 118, 96–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.M.; Knight, J. Introduction to genetic association studies. Cold Spring Harb. Protoc. 2012, 2012, 297–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birney, E. The International Human Genome Project. Hum. Mol. Genet. 2021, 30, R161–R163. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.G.; Choi, E.H.; Foster, C.B.; Chanock, S.J. Using genetic variation to study human disease. Trends Mol. Med. 2001, 7, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, C.S.; Penrod, N.M.; Williams, S.M.; Moore, J.H. Failure to replicate a genetic association may provide important clues about genetic architecture. PLoS ONE 2009, 4, e5639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsa, A. Genotype-phenotype correlations: Filling the void. Clin. J. Am. Soc. Nephrol. 2010, 5, 1542–1543. [Google Scholar] [CrossRef] [Green Version]
- Hirschhorn, J.N.; Lohmueller, K.; Byrne, E.; Hirschhorn, K. A comprehensive review of genetic association studies. Genet. Med. 2002, 4, 45–61. [Google Scholar] [CrossRef] [Green Version]
- Hattersley, A.T.; McCarthy, M.I. What makes a good genetic association study? Lancet 2005, 366, 1315–1323. [Google Scholar] [CrossRef]
- Sham, P.C.; Purcell, S.M. Statistical power and significance testing in large-scale genetic studies. Nat. Rev. Genet. 2014, 15, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Lenth, R.V. Some Practical Guidelines for Effective Sample Size Determination. Am. Stat. 2001, 55, 187–193. [Google Scholar] [CrossRef]
- Jones, S.R.; Carley, S.; Harrison, M. An introduction to power and sample size estimation. Emerg. Med. J. 2003, 20, 453–458. [Google Scholar] [CrossRef]
- Columb, M.O.; Atkinson, M.S. Statistical analysis: Sampe size and power estimations. BJA Educ. 2016, 16, 159–161. [Google Scholar] [CrossRef] [Green Version]
- Serdar, C.C.; Cihan, M.; Yücel, D.; Serdar, M.A. Sample size, power and effect size revisited: Simplified and practical approaches in pre-clinical, clinical and laboratory studies. Biochem. Med. 2021, 31, 010502. [Google Scholar] [CrossRef]
- Ioannidis, J.P. Genetic associations: False or true? Trends Mol. Med. 2003, 9, 135–138. [Google Scholar] [CrossRef]
- Hedrick, P.W. Genetics of Populations; Jones & Bartlett Learning Publishers: Burlington, MA, USA, 2011; p. 104. [Google Scholar]
- Shastry, B.S. SNPs in disease gene mapping, medicinal drug development and evolution. J. Hum. Genet. 2007, 52, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Gail, M.H.; Weinberg, C.R.; Carroll, R.J.; Chung, C.C.; Wang, Z.; Chanock, S.J.; Fraumeni, J.F., Jr.; Chatterjee, N. Distribution of allele frequencies and effect sizes and their interrelationships for common genetic susceptibility variants. Proc. Natl. Acad. Sci. USA 2011, 108, 18026–18031. [Google Scholar] [CrossRef] [Green Version]
- Goddard, K.A.; Hopkins, P.J.; Hall, J.M.; Witte, J.S. Linkage disequilibrium and allele-frequency distributions for 114 single-nucleotide polymorphisms in five populations. Am. J. Hum. Genet. 2000, 66, 216–234. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Przeworski, M. Linkage disequilibrium in humans: Models and data. Am. J. Hum. Genet. 2001, 69, 1–14. [Google Scholar] [CrossRef]
- Gaye, A.; Davis, S.K. Genetic model misspecification in genetic association studies. BMC Res. Notes 2017, 10, 569. [Google Scholar] [CrossRef] [Green Version]
- Burton, P.R.; Hansell, A.L.; Fortier, I.; Manolio, T.A.; Khoury, M.J.; Little, J.; Elliott, P. Size matters: Just how big is BIG?: Quantifying realistic sample size requirements for human genome epidemiology. Int. J. Epidemiol. 2009, 38, 263–273. [Google Scholar] [CrossRef]
- So, H.C.; Sham, P.C. Robust association tests under different genetic models, allowing for binary or quantitative traits and covariates. Behav. Genet. 2011, 41, 768–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, D.; Finch, S.J. Factors affecting statistical power in the detection of genetic association. J. Clin. Investig. 2005, 115, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.Y.; Day, N.E.; Luan, J.A.; Chan, K.P.; Wareham, N.J. The detection of gene-environment interaction for continuous traits: Should we deal with measurement error by bigger studies or better measurement? Int. J. Epidemiol. 2003, 32, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Zhao, H.; Millward, B.A.; Demaine, A.G. The Rate of Decline of Glomerular Filtration Rate May Not Be Associated with Polymorphism of the PPARγ2 Gene in Patients with Type 1 Diabetes and Nephropathy. PPAR Res. 2014, 2014, 523584. [Google Scholar] [CrossRef] [Green Version]
- Noordzij, M.; Dekker, F.W.; Zoccali, C.; Jager, K.J. Sample size calculations. Nephron Clin. Pract. 2011, 118, c319–c323. [Google Scholar] [CrossRef] [Green Version]
- Marder, B.; Schröppel, B.; Murphy, B. Genetic variability and transplantation. Curr. Opin. Urol. 2003, 13, 81–89. [Google Scholar] [CrossRef]
- Scalzotto, E.; Corradi, V.; Salin, A.; Caprara, C.; Skoumal, R.; Neri, A.; Cannone, M.; Frigo, A.; Chiaramonte, S.; Ferrari, F.; et al. Single Nucleotide Polymorphism Profiles of Patients with Acute Renal Rejection to Personalize Immunosuppressive Therapy: Preliminary Results from An On-Going, Italian Study. J. Organ Transplant. 2017, 1, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Neri, A.; Scalzotto, E.; Corradi, V.; Caprara, C.; Salin, A.; Cannone, M.; De Cal, M.; Romano, G.; Tulissi, P.; Cussigh, A.R.; et al. Acute rejection in kidney transplantation and the evaluation of associated polymorphisms (SNPs): The importance of sample size. Diagnosis 2019, 6, 287–295. [Google Scholar] [CrossRef]
- Lundberg, J.M.; Franco-Cereceda, A.; Lacroix, J.S.; Pernow, J. Neuropeptide Y and sympathetic neurotransmission. Ann. New York Acad. Sci. 1990, 611, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, A.; Michel, M.C. Renal effects of neuropeptide Y. Pflugers Arch. 1998, 435, 443–453. [Google Scholar] [CrossRef] [PubMed]
- White, J.D. Neuropeptide Y: A central regulator of energy homeostasis. Regul. Pept. 1993, 49, 93–107. [Google Scholar] [CrossRef]
- Karvonen, M.K.; Pesonen, U.; Koulu, M.; Niskanen, L.; Laakso, M.; Rissanen, A.; Dekker, J.M.; Hart, L.M.; Valve, R.; Uusitupa, M.I. Association of a leucine(7)-to-proline(7) polymorphism in the signal peptide of neuropeptide Y with high serum cholesterol and LDL cholesterol levels. Nat. Med. 1998, 4, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Kallio, J.; Pesonen, U.; Karvonen, M.K.; Kojima, M.; Hosoda, H.; Kangawa, K.; Koulu, M. Enhanced exercise-induced GH secretion in subjects with Pro7 substitution in the prepro-NPY. J. Clin. Endocrinol. Metab. 2001, 86, 5348–5352. [Google Scholar] [CrossRef] [PubMed]
- Ding, B. Distribution of the NPY 1128C allele frequency in different populations. J. Neural Transm. 2003, 110, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Nordman, S.; Möllsten, A.; Falhammar, H.; Brismar, K.; Dahlquist, G.; Efendic, S.; Gu, H.F. Distribution of neuropeptide Y Leu7Pro polymorphism in patients with type 1 diabetes and diabetic nephropathy among Swedish and American populations. Eur. J. Endocrinol. 2007, 157, 641–645. [Google Scholar] [CrossRef] [Green Version]
- Gauderman, W.J. Sample size requirements for matched case-control studies of gene-environment interaction. Stat. Med. 2002, 21, 35–50. [Google Scholar] [CrossRef]
- Kang, M.; Choi, S.; Koh, I. The effect of Increasing control-to-case ratio on statistical power in a simulated case-control SNP association study. Genom. Inform. 2009, 7, 148–151. [Google Scholar] [CrossRef] [Green Version]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaye, A.; Burton, T.W.; Burton, P.R. ESPRESSO: Taking into account assessment errors on outcome and exposures in power analysis for association studies. Bioinformatics 2015, 31, 2691–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, C.M.; Jacobson, S.A.; Fingerlin, T.E. Power and Sample Size Calculations for Genetic Association Studies in the Presence of Genetic Model Misspecification. Hum. Hered. 2019, 84, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Finch, S.J.; Nothnagel, M.; Ott, J. Power and sample size calculations for case-control genetic association tests when errors are present: Application to single nucleotide polymorphisms. Hum. Hered. 2002, 54, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Cherny, S.S.; Sham, P.C. Genetic Power Calculator: Design of linkage and association genetic mapping studies of complex traits. Bioinformatics 2003, 19, 149–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menashe, I.; Rosenberg, P.S.; Chen, B.E. PGA: Power calculator for case-control genetic association analyses. BMC Genet. 2008, 9, 36–40. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Definition | ||
|---|---|---|
| Basic | Alpha (Type I error or significance level) | The probability of incorrectly rejecting H0 according to sample results when it is actually true in the source population (false positive). The conventional value of the significance level is set at 0.05 indicating that there is a 5% probability of incorrectly rejecting the true H0. |
| Beta (Type II error) | The probability of incorrectly retaining H0 according to sample results when it is false in the source population (false negative). The conventional value of this second kind of error is set at 0.20, meaning that the probability of failing to detect a statistically significant difference when it does exist, is no more than 20%. | |
| Power (1-beta) | The probability of correctly detecting a significant difference in the sample when it actually exists in the source population. Being the complement of beta, the conventional value of the power is 0.80, indicating that there is 80% probability of avoiding a false negative result. | |
| Genetic | Prevalence | The proportion of individuals with the disease in a population at a given point in time. The smaller the proportion of affected subjects, the larger the sample size required. |
| Effect size | The numeric value that indicates the strength of the association between a gene variant and a disease or the magnitude of the difference in the allelic/genotypic frequency between groups (i.e., cases and controls). For binary outcomes, it is expressed as a relative risk/odds ratio, while for continuous outcomes, as a percentage of the phenotypic variance. The smaller the effect size, the larger the sample size required. | |
| Minor allele frequency (MAF) | The frequency of the second most frequent allele for a polymorphism. According to MAF, a genetic variant can be classified as common (MAF > 5%), uncommon (1–5% MAF) and rare (MAF < 1%). The higher the MAF, the smaller the number of patients to be studied. | |
| Linkage disequilibrium (LD) | The non-random association of two or more alleles at different loci that occur together on the same haplotype more often than expected. The stronger the linkage between the marker allele and the disease allele, the higher the power of the study and the lower the sample size required to detect the gene–disease association. | |
| Genetic inheritance model | The model of inheritance of a genetic variant. Three genetic models are usually adopted: dominant, recessive and additive model. Genetic model misspecification reduces the study power and thus requires a higher sample size. | |
| Errors | They result from genotype/phenotype misclassification and/or missing genotype data. Their effect on the power loss cannot be quantified at the design stage of the study. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Politi, C.; Roumeliotis, S.; Tripepi, G.; Spoto, B. Sample Size Calculation in Genetic Association Studies: A Practical Approach. Life 2023, 13, 235. https://doi.org/10.3390/life13010235
Politi C, Roumeliotis S, Tripepi G, Spoto B. Sample Size Calculation in Genetic Association Studies: A Practical Approach. Life. 2023; 13(1):235. https://doi.org/10.3390/life13010235
Chicago/Turabian StylePoliti, Cristina, Stefanos Roumeliotis, Giovanni Tripepi, and Belinda Spoto. 2023. "Sample Size Calculation in Genetic Association Studies: A Practical Approach" Life 13, no. 1: 235. https://doi.org/10.3390/life13010235
APA StylePoliti, C., Roumeliotis, S., Tripepi, G., & Spoto, B. (2023). Sample Size Calculation in Genetic Association Studies: A Practical Approach. Life, 13(1), 235. https://doi.org/10.3390/life13010235