
Sodium–Glucose Transporter 2 (SGLT2) Inhibitors and Iron Deficiency in Heart Failure and Chronic Kidney Disease: A Literature Review

 ,
,  and
and 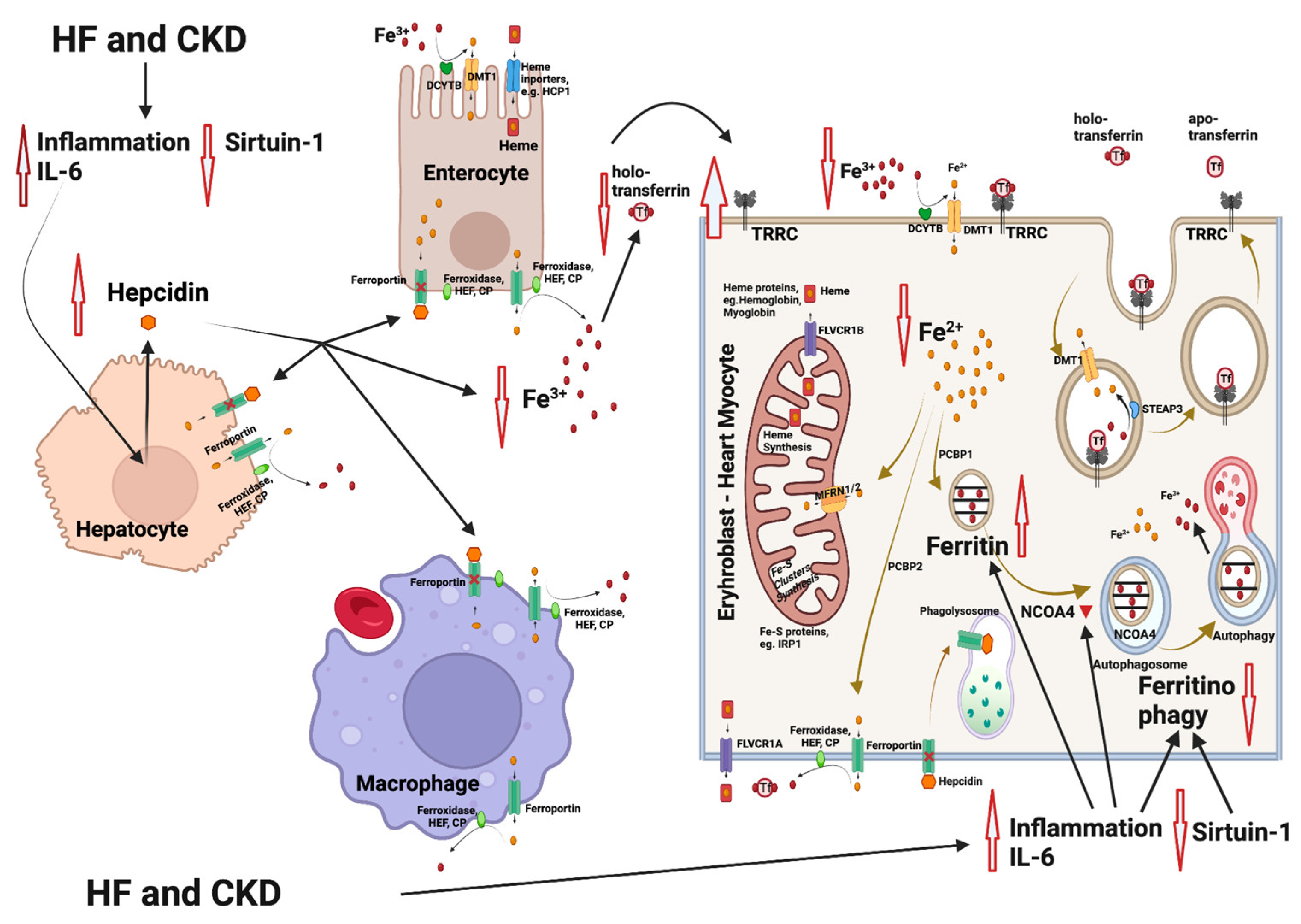{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Effects of SGLT2 Inhibitors
3. Iron Metabolism
4. Iron Deficiency Anemia in Heart Failure and Chronic Kidney Disease
5. Influence of SGLT2 Inhibitors on Erythropoiesis and Iron Homeostasis
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.K.L.; Pinsky, J.L.; Kannel, W.B.; Levy, D. The Epidemiology of Heart Failure: The Framingham Study. J. Am. Coll. Cardiol. 1993, 22, A6–A13. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Normand, S.-L.T.; Wang, Y.; Krumholz, H.M. National and Regional Trends in Heart Failure Hospitalization and Mortality Rates for Medicare Beneficiaries, 1998–2008. JAMA 2011, 306, 1669. [Google Scholar] [CrossRef] [PubMed]
- Bragazzi, N.L.; Zhong, W.; Shu, J.; Abu Much, A.; Lotan, D.; Grupper, A.; Younis, A.; Dai, H. Burden of Heart Failure and Underlying Causes in 195 Countries and Territories from 1990 to 2017. Eur. J. Prev. Cardiol. 2021, 28, 1682–1690. [Google Scholar] [CrossRef] [PubMed]
- Agbor, V.N.; Essouma, M.; Ntusi, N.A.B.; Nyaga, U.F.; Bigna, J.J.; Noubiap, J.J. Heart Failure in Sub-Saharan Africa: A Contemporaneous Systematic Review and Meta-Analysis. Int. J. Cardiol. 2018, 257, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Soenarta, A.A.; Buranakitjaroen, P.; Chia, Y.-C.; Chen, C.-H.; Nailes, J.; Hoshide, S.; Minh, H.V.; Park, S.; Shin, J.; Siddique, S.; et al. An Overview of Hypertension and Cardiac Involvement in Asia: Focus on Heart Failure. J. Clin. Hypertens. 2020, 22, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Kovesdy, C.P. Epidemiology of Chronic Kidney Disease: An Update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef]
- Triposkiadis, F.K.; Skoularigis, J. Prevalence and Importance of Comorbidities in Patients with Heart Failure. Curr. Heart Fail. Rep. 2012, 9, 354–362. [Google Scholar] [CrossRef]
- Yang, Y.; Huang, Z.; Wu, B.; Lu, J.; Xiu, J.; Tu, J.; Chen, S.; Pan, Y.; Bao, K.; Wang, J.; et al. Predictors of Mortality in Heart Failure with Reduced Ejection Fraction: Interaction between Diabetes Mellitus and Impaired Renal Function. Int. Urol. Nephrol. 2023, 55, 2285–2293. [Google Scholar] [CrossRef]
- Conrad, N.; Judge, A.; Tran, J.; Mohseni, H.; Hedgecott, D.; Crespillo, A.P.; Allison, M.; Hemingway, H.; Cleland, J.G.; McMurray, J.J.V.; et al. Temporal Trends and Patterns in Heart Failure Incidence: A Population-Based Study of 4 Million Individuals. Lancet 2018, 391, 572–580. [Google Scholar] [CrossRef]
- Tuegel, C.; Bansal, N. Heart Failure in Patients with Kidney Disease. Heart 2017, 103, 1848–1853. [Google Scholar] [CrossRef] [PubMed]
- Anand, I.S.; Gupta, P. Anemia and Iron Deficiency in Heart Failure: Current Concepts and Emerging Therapies. Circulation 2018, 138, 80–98. [Google Scholar] [CrossRef] [PubMed]
- Babitt, J.L.; Lin, H.Y. Mechanisms of Anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634. [Google Scholar] [CrossRef] [PubMed]
- Fishbane, S.; Pollack, S.; Feldman, H.I.; Joffe, M.M. Iron Indices in Chronic Kidney Disease in the National Health and Nutritional Examination Survey 1988–2004. Clin. J. Am. Soc. Nephrol. 2009, 4, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, E.K.; Kapitsinou, P.; Pergola, P.E.; Kovesdy, C.P.; Jalal, D.I. Iron Deficiency in Chronic Kidney Disease: Updates on Pathophysiology, Diagnosis, and Treatment. J. Am. Soc. Nephrol. 2020, 31, 456–468. [Google Scholar] [CrossRef]
- Kristensen, S.L.; Rørth, R.; Jhund, P.S.; Docherty, K.F.; Sattar, N.; Preiss, D.; Køber, L.; Petrie, M.C.; McMurray, J.J.V. Cardiovascular, Mortality, and Kidney Outcomes with GLP-1 Receptor Agonists in Patients with Type 2 Diabetes: A Systematic Review and Meta-Analysis of Cardiovascular Outcome Trials. Lancet Diabetes Endocrinol. 2019, 7, 776–785. [Google Scholar] [CrossRef]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 Inhibitors for Primary and Secondary Prevention of Cardiovascular and Renal Outcomes in Type 2 Diabetes: A Systematic Review and Meta-Analysis of Cardiovascular Outcome Trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Stefánsson, B.V.; Heerspink, H.J.L.; Wheeler, D.C.; Sjöström, C.D.; Greasley, P.J.; Sartipy, P.; Cain, V.; Correa-Rotter, R. Correction of Anemia by Dapagliflozin in Patients with Type 2 Diabetes. J. Diabetes Complicat. 2020, 34, 107729. [Google Scholar] [CrossRef]
- Oshima, M.; Neuen, B.L.; Jardine, M.J.; Bakris, G.; Edwards, R.; Levin, A.; Mahaffey, K.W.; Neal, B.; Pollock, C.; Rosenthal, N.; et al. Effects of Canagliflozin on Anaemia in Patients with Type 2 Diabetes and Chronic Kidney Disease: A Post-Hoc Analysis from the CREDENCE Trial. Lancet Diabetes Endocrinol. 2020, 8, 903–914. [Google Scholar] [CrossRef]
- Docherty, K.F.; Welsh, P.; Verma, S.; De Boer, R.A.; O’Meara, E.; Bengtsson, O.; Køber, L.; Kosiborod, M.N.; Hammarstedt, A.; Langkilde, A.M.; et al. Iron Deficiency in Heart Failure and Effect of Dapagliflozin: Findings From DAPA-HF. Circulation 2022, 146, 980–994. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Yao, L.; Liu, X.; Xu, T.; Tian, B. Effects of Sodium-Glucose Co-Transporter 2 Inhibitors on Hemoglobin Levels: A Meta-Analysis of Randomized Controlled Trials. Front. Pharmacol. 2021, 12, 630820. [Google Scholar] [CrossRef] [PubMed]
- Kanbay, M.; Tapoi, L.; Ureche, C.; Tanriover, C.; Cevik, E.; Demiray, A.; Afsar, B.; Cherney, D.Z.I.; Covic, A. Effect of Sodium-Glucose Cotransporter 2 Inhibitors on Hemoglobin and Hematocrit Levels in Type 2 Diabetes: A Systematic Review and Meta-Analysis. Int. Urol. Nephrol. 2022, 54, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Portolés, J.; Martín, L.; Broseta, J.J.; Cases, A. Anemia in Chronic Kidney Disease: From Pathophysiology and Current Treatments, to Future Agents. Front. Med. 2021, 8, 642296. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Goto, S. Possible Mechanism of Hematocrit Elevation by Sodium Glucose Cotransporter 2 Inhibitors and Associated Beneficial Renal and Cardiovascular Effects. Circulation 2019, 139, 1985–1987. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Zinman, B.; Fitchett, D.; Wanner, C.; Ferrannini, E.; Schumacher, M.; Schmoor, C.; Ohneberg, K.; Johansen, O.E.; George, J.T.; et al. How Does Empagliflozin Reduce Cardiovascular Mortality? Insights From a Mediation Analysis of the EMPA-REG OUTCOME Trial. Diabetes Care 2017, 41, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zhu, Y.; Zhao, W.; Wu, M.; Huang, H.; Huang, H.; Wu, C.; Zhou, X.; Zhou, S.; Wang, C.; et al. Rationale and Design of the ADIDAS Study: Association Between Dapagliflozin-Induced Improvement and Anemia in Heart Failure Patients. Cardiovasc. Drugs Ther. 2022, 36, 505–509. [Google Scholar] [CrossRef]
- Vergara, A.; Jacobs-Cachá, C.; Soler, M.J. Sodium-Glucose Cotransporter Inhibitors: Beyond Glycaemic Control. Clin. Kidney J. 2019, 12, 322–325. [Google Scholar] [CrossRef]
- Wanner, C.; Heerspink, H.J.L.; Zinman, B.; Inzucchi, S.E.; Koitka-Weber, A.; Mattheus, M.; Hantel, S.; Woerle, H.-J.; Broedl, U.C.; von Eynatten, M.; et al. Empagliflozin and Kidney Function Decline in Patients with Type 2 Diabetes: A Slope Analysis from the EMPA-REG OUTCOME Trial. J. Am. Soc. Nephrol. 2018, 29, 2755–2769. [Google Scholar] [CrossRef]
- Kidokoro, K.; Cherney, D.Z.I.; Bozovic, A.; Nagasu, H.; Satoh, M.; Kanda, E.; Sasaki, T.; Kashihara, N. Evaluation of Glomerular Hemodynamic Function by Empagliflozin in Diabetic Mice Using In Vivo Imaging. Circulation 2019, 140, 303–315. [Google Scholar] [CrossRef]
- Wanner, C. Sodium Glucose Cotransporter 2 Inhibition and the Visualization of Kidney Hemodynamics. Circulation 2019, 140, 316–318. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, C.C.J.; Petrykiv, S.; Laverman, G.D.; Cherney, D.Z.; Gansevoort, R.T.; Heerspink, H.J.L. Effects of the SGLT-2 Inhibitor Dapagliflozin on Glomerular and Tubular Injury Markers. Diabetes Obes. Metab. 2018, 20, 1988–1993. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Desai, M.; Jardine, M.; Balis, D.; Meininger, G.; Perkovic, V. Canagliflozin Slows Progression of Renal Function Decline Independently of Glycemic Effects. J. Am. Soc. Nephrol. 2017, 28, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Satirapoj, B.; Korkiatpitak, P.; Supasyndh, O. Effect of Sodium-Glucose Cotransporter 2 Inhibitor on Proximal Tubular Function and Injury in Patients with Type 2 Diabetes: A Randomized Controlled Trial. Clin. Kidney J. 2019, 12, 326–332. [Google Scholar] [CrossRef]
- Bolinder, J.; Ljunggren, Ö.; Kullberg, J.; Johansson, L.; Wilding, J.; Langkilde, A.M.; Sugg, J.; Parikh, S. Effects of Dapagliflozin on Body Weight, Total Fat Mass, and Regional Adipose Tissue Distribution in Patients with Type 2 Diabetes Mellitus with Inadequate Glycemic Control on Metformin. J. Clin. Endocrinol. Metab. 2012, 97, 1020–1031. [Google Scholar] [CrossRef]
- Sato, T.; Aizawa, Y.; Yuasa, S.; Kishi, S.; Fuse, K.; Fujita, S.; Ikeda, Y.; Kitazawa, H.; Takahashi, M.; Sato, M.; et al. The Effect of Dapagliflozin Treatment on Epicardial Adipose Tissue Volume. Cardiovasc. Diabetol. 2018, 17, 6. [Google Scholar] [CrossRef]
- Mulder, S.; Heerspink, H.J.L.; Darshi, M.; Kim, J.J.; Laverman, G.D.; Sharma, K.; Pena, M.J. Effects of Dapagliflozin on Urinary Metabolites in People with Type 2 Diabetes. Diabetes Obes. Metab. 2019, 21, 2422–2428. [Google Scholar] [CrossRef]
- Birnbaum, Y.; Bajaj, M.; Yang, H.-C.; Ye, Y. Combined SGLT2 and DPP4 Inhibition Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Nephropathy in Mice with Type 2 Diabetes. Cardiovasc. Drugs Ther. 2018, 32, 135–145. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin Reduces Inflammation and Fibrosis Biomarkers: A Potential Mechanism of Action for Beneficial Effects of SGLT2 Inhibitors in Diabetic Kidney Disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, S.H.; Kang, J.M.; Heo, J.H.; Kim, D.-J.; Park, S.H.; Sung, M.; Kim, J.; Oh, J.; Yang, D.H.; et al. Empagliflozin Attenuates Diabetic Tubulopathy by Improving Mitochondrial Fragmentation and Autophagy. Am. J. Physiol. Renal. Physiol. 2019, 317, F767–F780. [Google Scholar] [CrossRef] [PubMed]
- Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; Ng, S.Y.A.; et al. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Zelniker, T.A.; Braunwald, E. Mechanisms of Cardiorenal Effects of Sodium-Glucose Cotransporter 2 Inhibitors: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Lehrke, M. SGLT2 Inhibition: Changing What Fuels the Heart. J. Am. Coll. Cardiol. 2019, 73, 1945–1947. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Takashima, H.; Oguma, H.; Nakamura, Y.; Ohno, M.; Utsunomiya, K.; Furukawa, T.; Tei, R.; Abe, M. Canagliflozin Improves Erythropoiesis in Diabetes Patients with Anemia of Chronic Kidney Disease. Diabetes Technol. Ther. 2019, 21, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Zelniker, T.A.; Braunwald, E. Clinical Benefit of Cardiorenal Effects of Sodium-Glucose Cotransporter 2 Inhibitors: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, B.; Garrick, M.D. Iron Imports. II. Iron Uptake at the Apical Membrane in the Intestine. Am. J. Physiol.-Gastrointest. Liver Physiol. 2005, 289, G981–G986. [Google Scholar] [CrossRef] [PubMed]
- Yanatori, I.; Kishi, F. DMT1 and Iron Transport. Free Radic. Biol. Med. 2019, 133, 55–63. [Google Scholar] [CrossRef]
- Zhang, X.-D.; Liu, Z.-Y.; Wang, M.-S.; Guo, Y.-X.; Wang, X.-K.; Luo, K.; Huang, S.; Li, R.-F. Mechanisms and Regulations of Ferroptosis. Front. Immunol. 2023, 14, 1269451. [Google Scholar] [CrossRef]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef]
- Nemeth, E.; Ganz, T. Hepcidin-Ferroportin Interaction Controls Systemic Iron Homeostasis. Int. J. Mol. Sci. 2021, 22, 6493. [Google Scholar] [CrossRef] [PubMed]
- Dev, S.; Babitt, J.L. Overview of Iron Metabolism in Health and Disease. Hemodial. Int. 2017, 21 (Suppl. S1), S6–S20. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C.; Schmidt, P.J. Iron Homeostasis. Annu. Rev. Physiol. 2007, 69, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Singer, C.E.; Vasile, C.M.; Popescu, M.; Popescu, A.I.S.; Marginean, I.C.; Iacob, G.A.; Popescu, M.D.; Marginean, C.M. Role of Iron Deficiency in Heart Failure-Clinical and Treatment Approach: An Overview. Diagnostics 2023, 13, 304. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. How Can Sodium-Glucose Cotransporter 2 Inhibitors Stimulate Erythrocytosis in Patients Who Are Iron-Deficient? Implications for Understanding Iron Homeostasis in Heart Failure. Eur. J. Heart Fail. 2022, 24, 2287–2296. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Pagani, A. Advances in Understanding Iron Metabolism and Its Crosstalk with Erythropoiesis. Br. J. Haematol. 2018, 182, 481–494. [Google Scholar] [CrossRef]
- Nemeth, E.; Valore, E.V.; Territo, M.; Schiller, G.; Lichtenstein, A.; Ganz, T. Hepcidin, a Putative Mediator of Anemia of Inflammation, Is a Type II Acute-Phase Protein. Blood 2003, 101, 2461–2463. [Google Scholar] [CrossRef]
- Zaritsky, J.; Young, B.; Wang, H.-J.; Westerman, M.; Olbina, G.; Nemeth, E.; Ganz, T.; Rivera, S.; Nissenson, A.R.; Salusky, I.B. Hepcidin--a Potential Novel Biomarker for Iron Status in Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2009, 4, 1051–1056. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Liakopoulos, V.; Antoniadi, G.; Kartsios, C.; Stefanidis, I. The Role of Hepcidin in Iron Homeostasis and Anemia in Hemodialysis Patients. Semin. Dial. 2009, 22, 70–77. [Google Scholar] [CrossRef]
- Ashby, D.R.; Gale, D.P.; Busbridge, M.; Murphy, K.G.; Duncan, N.D.; Cairns, T.D.; Taube, D.H.; Bloom, S.R.; Tam, F.W.K.; Chapman, R.S.; et al. Plasma Hepcidin Levels Are Elevated but Responsive to Erythropoietin Therapy in Renal Disease. Kidney Int. 2009, 75, 976–981. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Iron Balance and the Role of Hepcidin in Chronic Kidney Disease. Semin. Nephrol. 2016, 36, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Ku, E.; Del Vecchio, L.; Eckardt, K.-U.; Haase, V.H.; Johansen, K.L.; Nangaku, M.; Tangri, N.; Waikar, S.S.; Więcek, A.; Cheung, M.; et al. Novel Anemia Therapies in Chronic Kidney Disease: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2023, 104, 655–680. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Burdmann, E.A.; Chen, C.-Y.; Cooper, M.E.; De Zeeuw, D.; Eckardt, K.-U.; Feyzi, J.M.; Ivanovich, P.; Kewalramani, R.; Levey, A.S.; et al. A Trial of Darbepoetin Alfa in Type 2 Diabetes and Chronic Kidney Disease. N. Engl. J. Med. 2009, 361, 2019–2032. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Kirwan, B.-A.; van Veldhuisen, D.J.; Filippatos, G.; Comin-Colet, J.; Ruschitzka, F.; Lüscher, T.F.; Arutyunov, G.P.; Motro, M.; Mori, C.; et al. Effects of Ferric Carboxymaltose on Hospitalisations and Mortality Rates in Iron-Deficient Heart Failure Patients: An Individual Patient Data Meta-Analysis. Eur. J. Heart Fail. 2018, 20, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Yamani, N.; Ahmed, A.; Gosain, P.; Fatima, K.; Shaikh, A.T.; Qamar, H.; Shahid, I.; Arshad, M.S.; Almas, T.; Figueredo, V. Effect of Iron Supplementation in Patients with Heart Failure and Iron Deficiency: A Systematic Review and Meta-Analysis. IJC Heart Vasc. 2021, 36, 100871. [Google Scholar] [CrossRef] [PubMed]
- Awan, A.A.; Walther, C.P.; Richardson, P.A.; Shah, M.; Winkelmayer, W.C.; Navaneethan, S.D. Prevalence, Correlates and Outcomes of Absolute and Functional Iron Deficiency Anemia in Nondialysis-Dependent Chronic Kidney Disease. Nephrol. Dial. Transplant. 2021, 36, 129–136. [Google Scholar] [CrossRef]
- Macdougall, I.C.; White, C.; Anker, S.D.; Bhandari, S.; Farrington, K.; Kalra, P.A.; McMurray, J.J.V.; Murray, H.; Tomson, C.R.V.; Wheeler, D.C.; et al. Intravenous Iron in Patients Undergoing Maintenance Hemodialysis. N. Engl. J. Med. 2019, 380, 447–458. [Google Scholar] [CrossRef]
- Mazer, C.D.; Hare, G.M.T.; Connelly, P.W.; Gilbert, R.E.; Shehata, N.; Quan, A.; Teoh, H.; Leiter, L.A.; Zinman, B.; Jüni, P.; et al. Effect of Empagliflozin on Erythropoietin Levels, Iron Stores, and Red Blood Cell Morphology in Patients with Type 2 Diabetes Mellitus and Coronary Artery Disease. Circulation 2020, 141, 704–707. [Google Scholar] [CrossRef]
- Cases, A.; Górriz, J.L.; Cigarrán, S.; Nuñez, J. Efecto de Los Inhibidores Del Cotransportador Sodio-Glucosa Tipo 2 Sobre La Anemia: Posibles Implicaciones Clínicas. Nefrología 2023, in press. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; de Zeeuw, D.; Wie, L.; Leslie, B.; List, J. Dapagliflozin a Glucose-Regulating Drug with Diuretic Properties in Subjects with Type 2 Diabetes. Diabetes Obes. Metab. 2013, 15, 853–862. [Google Scholar] [CrossRef]
- O’Neill, J.; Fasching, A.; Pihl, L.; Patinha, D.; Franzén, S.; Palm, F. Acute SGLT Inhibition Normalizes O2 Tension in the Renal Cortex but Causes Hypoxia in the Renal Medulla in Anaesthetized Control and Diabetic Rats. Am. J. Physiol. Renal. Physiol. 2015, 309, F227–F234. [Google Scholar] [CrossRef]
- Packer, M. Mutual Antagonism of Hypoxia-Inducible Factor Isoforms in Cardiac, Vascular, and Renal Disorders. JACC Basic Transl. Sci. 2020, 5, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, K.-U.; Kurtz, A. Regulation of Erythropoietin Production. Eur. J. Clin. Investig. 2005, 35 (Suppl. S3), 13–19. [Google Scholar] [CrossRef] [PubMed]
- Fuchs Andersen, C.; Omar, M.; Glenthøj, A.; El Fassi, D.; Møller, H.J.; Lindholm Kurtzhals, J.A.; Styrishave, B.; Kistorp, C.; Tuxen, C.; Poulsen, M.K.; et al. Effects of Empagliflozin on Erythropoiesis in Heart Failure: Data from the Empire HF Trial. Eur. J. Heart Fail. 2023, 25, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Zannad, F.; Ferreira, J.P.; Butler, J.; Filippatos, G.; Januzzi, J.L.; Sumin, M.; Zwick, M.; Saadati, M.; Pocock, S.J.; Sattar, N.; et al. Effect of Empagliflozin on Circulating Proteomics in Heart Failure: Mechanistic Insights into the EMPEROR Programme. Eur. Heart J. 2022, 43, 4991–5002. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-H.; Lee, Y.-M.; Chun, Y.-S.; Chen, J.; Kim, J.-E.; Park, J.-W. Sirtuin 1 Modulates Cellular Responses to Hypoxia by Deacetylating Hypoxia-Inducible Factor 1α. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Horiuchi, T. Hypoxia-Inducible Factor-Prolyl-Hydroxylase and Sodium-Glucose Cotransporter 2 Inhibitors for Low-Risk Myelodysplastic Syndrome-Related Anemia in Patients with Chronic Kidney Disease: A Report of Three Cases. Hematol. Rep. 2023, 15, 180–187. [Google Scholar] [CrossRef]
- Ferrannini, E.; Murthy, A.C.; Lee, Y.-H.; Muscelli, E.; Weiss, S.; Ostroff, R.M.; Sattar, N.; Williams, S.A.; Ganz, P. Mechanisms of Sodium-Glucose Cotransporter 2 Inhibition: Insights from Large-Scale Proteomics. Diabetes Care 2020, 43, 2183–2189. [Google Scholar] [CrossRef]
- Ghanim, H.; Abuaysheh, S.; Hejna, J.; Green, K.; Batra, M.; Makdissi, A.; Chaudhuri, A.; Dandona, P. Dapagliflozin Suppresses Hepcidin and Increases Erythropoiesis. J. Clin. Endocrinol. Metab. 2020, 105, dgaa057. [Google Scholar] [CrossRef]
- Packer, M. Role of Deranged Energy Deprivation Signaling in the Pathogenesis of Cardiac and Renal Disease in States of Perceived Nutrient Overabundance. Circulation 2020, 141, 2095–2105. [Google Scholar] [CrossRef]
- Li, D.; Liu, X.; Pi, W.; Zhang, Y.; Yu, L.; Xu, C.; Sun, Z.; Jiang, J. Fisetin Attenuates Doxorubicin-Induced Cardiomyopathy In Vivo and In Vitro by Inhibiting Ferroptosis Through SIRT1/Nrf2 Signaling Pathway Activation. Front. Pharmacol. 2022, 12, 808480. [Google Scholar] [CrossRef]
- Ma, S.; Sun, L.; Wu, W.; Wu, J.; Sun, Z.; Ren, J. USP22 Protects Against Myocardial Ischemia–Reperfusion Injury via the SIRT1-P53/SLC7A11–Dependent Inhibition of Ferroptosis–Induced Cardiomyocyte Death. Front. Physiol. 2020, 11, 551318. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Guo, J.; Huang, S.; Cheng, Y.; Luo, F.; Xu, X.; Chen, R.; Ma, G.; Wang, Y. The Roles of Sirtuins in Ferroptosis. Front. Physiol. 2023, 14, 660. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zheng, S.; Fan, Y.; Tan, K. Emerging Significance and Therapeutic Targets of Ferroptosis: A Potential Avenue for Human Kidney Diseases. Cell Death Dis. 2023, 14, 628. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Xia, Y.; Li, L.; Li, B.; Chen, L.; Yu, W.; Ruan, Y.; Rao, T.; Zhou, X.; Cheng, F. P53 Deacetylation Alleviates Calcium Oxalate Deposition-Induced Renal Fibrosis by Inhibiting Ferroptosis. Biomed. Pharmacother. 2023, 164, 114925. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Li, X.; Zhang, D.; Han, W. Astragaloside-IV Alleviates High Glucose-Induced Ferroptosis in Retinal Pigment Epithelial Cells by Disrupting the Expression of miR-138-5p/Sirt1/Nrf2. Bioengineered 2022, 13, 8238–8253. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, T.; Tong, Y.; Cui, R.; Qu, K.; Liu, C.; Zhang, J. Ulinastatin Protects against Acetaminophen-Induced Liver Injury by Alleviating Ferroptosis via the SIRT1/NRF2/HO-1 Pathway. Am. J. Transl. Res. 2021, 13, 6031–6042. [Google Scholar]
- Dang, R.; Wang, M.; Li, X.; Wang, H.; Liu, L.; Wu, Q.; Zhao, J.; Ji, P.; Zhong, L.; Licinio, J.; et al. Edaravone Ameliorates Depressive and Anxiety-like Behaviors via Sirt1/Nrf2/HO-1/Gpx4 Pathway. J. Neuroinflamm. 2022, 19, 41. [Google Scholar] [CrossRef]
- Liu, J.; Huang, J.; Zhang, Z.; Zhang, R.; Sun, Q.; Zhang, Z.; Liu, Y.; Ma, B. Mesenchymal Stem Cell-Derived Exosomes Ameliorate Delayed Neurocognitive Recovery in Aged Mice by Inhibiting Hippocampus Ferroptosis via Activating SIRT1/Nrf2/HO-1 Signaling Pathway. Oxidative Med. Cell Longev. 2022, 2022, 3593294. [Google Scholar] [CrossRef]
- Packer, M. Potential Interactions When Prescribing SGLT2 Inhibitors and Intravenous Iron in Combination in Heart Failure. JACC Heart Fail. 2023, 11, 106–114. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tziastoudi, M.; Pissas, G.; Golfinopoulos, S.; Filippidis, G.; Dousdampanis, P.; Eleftheriadis, T.; Stefanidis, I. Sodium–Glucose Transporter 2 (SGLT2) Inhibitors and Iron Deficiency in Heart Failure and Chronic Kidney Disease: A Literature Review. Life 2023, 13, 2338. https://doi.org/10.3390/life13122338
Tziastoudi M, Pissas G, Golfinopoulos S, Filippidis G, Dousdampanis P, Eleftheriadis T, Stefanidis I. Sodium–Glucose Transporter 2 (SGLT2) Inhibitors and Iron Deficiency in Heart Failure and Chronic Kidney Disease: A Literature Review. Life. 2023; 13(12):2338. https://doi.org/10.3390/life13122338
Chicago/Turabian StyleTziastoudi, Maria, Georgios Pissas, Spyridon Golfinopoulos, Georgios Filippidis, Periklis Dousdampanis, Theodoros Eleftheriadis, and Ioannis Stefanidis. 2023. "Sodium–Glucose Transporter 2 (SGLT2) Inhibitors and Iron Deficiency in Heart Failure and Chronic Kidney Disease: A Literature Review" Life 13, no. 12: 2338. https://doi.org/10.3390/life13122338
APA StyleTziastoudi, M., Pissas, G., Golfinopoulos, S., Filippidis, G., Dousdampanis, P., Eleftheriadis, T., & Stefanidis, I. (2023). Sodium–Glucose Transporter 2 (SGLT2) Inhibitors and Iron Deficiency in Heart Failure and Chronic Kidney Disease: A Literature Review. Life, 13(12), 2338. https://doi.org/10.3390/life13122338