
Mass Spectrometry-Based Proteomics of Minor Species in the Bulk: Questions to Raise with Respect to the Untargeted Analysis of Viral Proteins in Human Tissue



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
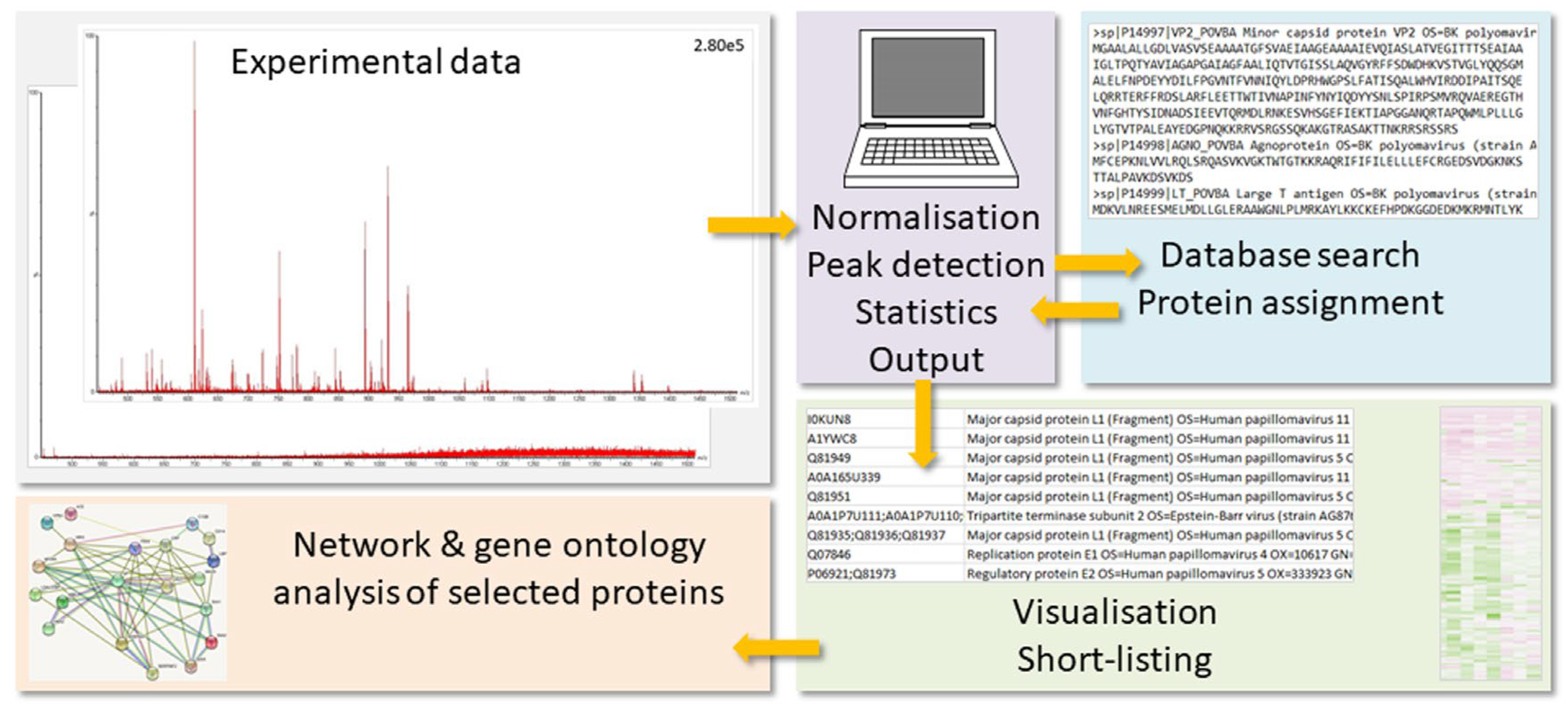
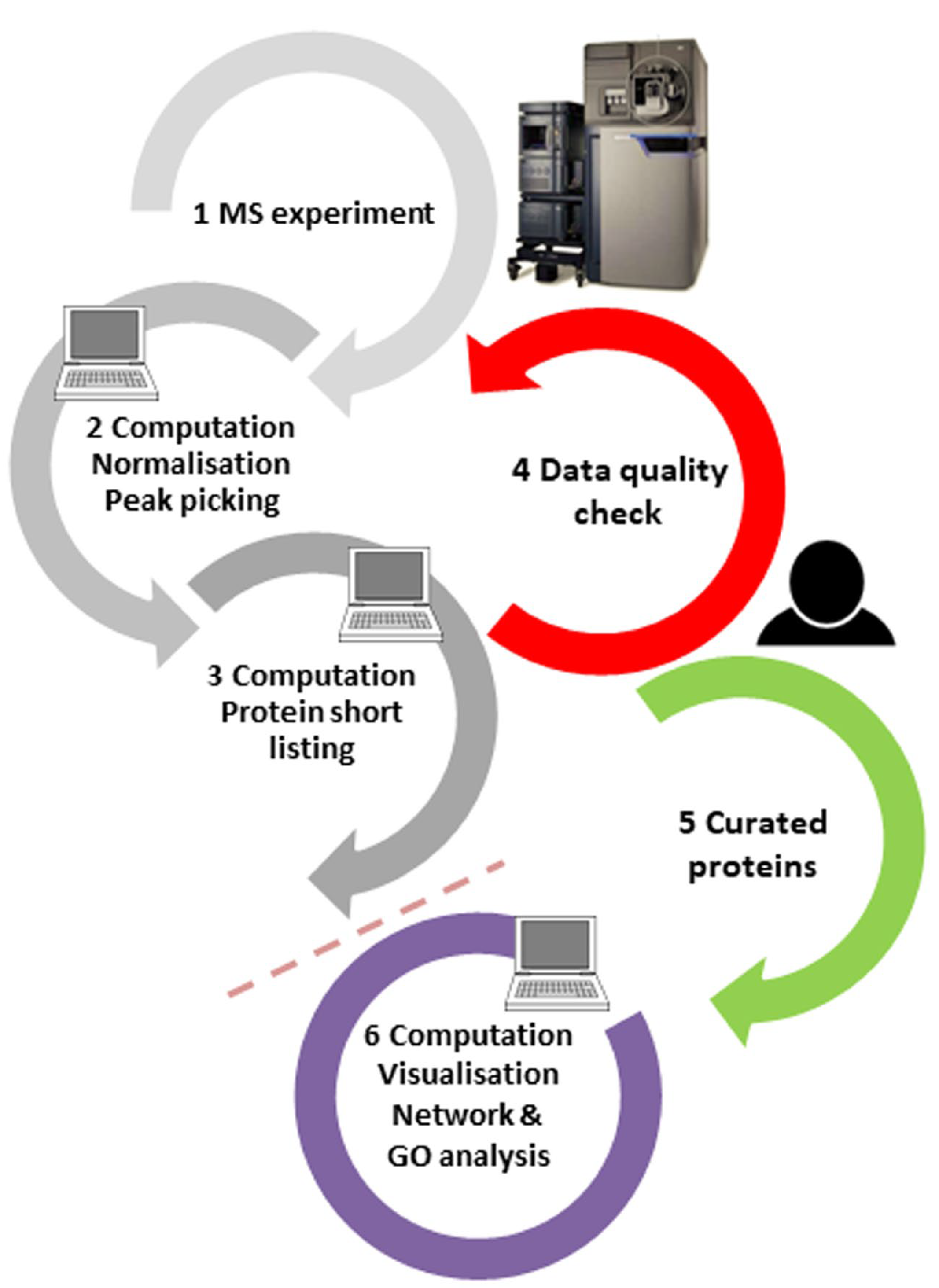
1. The Mass-Spectrometry-Based Proteomics Workflow and its Limits
2. Database Search
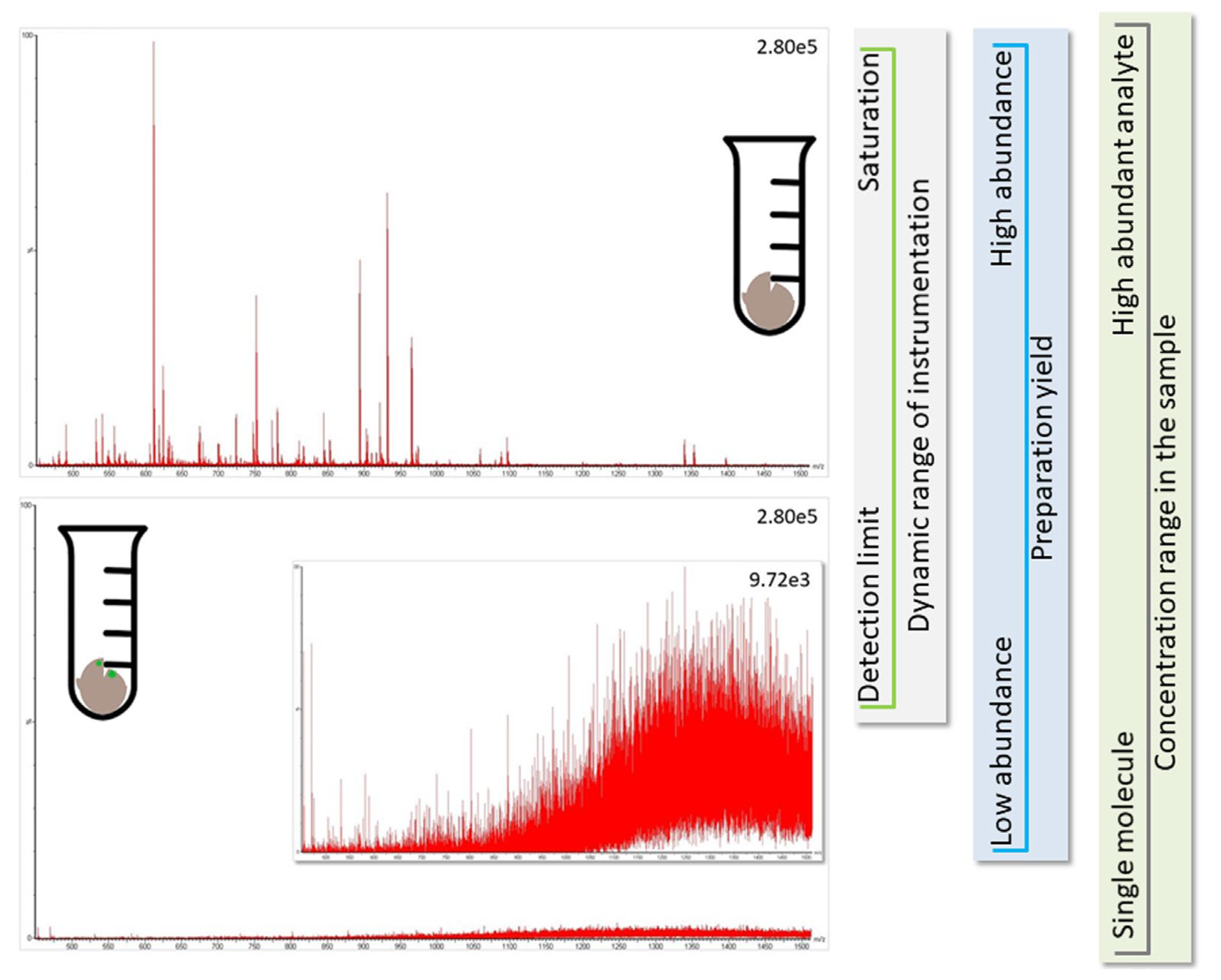
2.1. Proteomics of Minor Species
2.2. Viromics
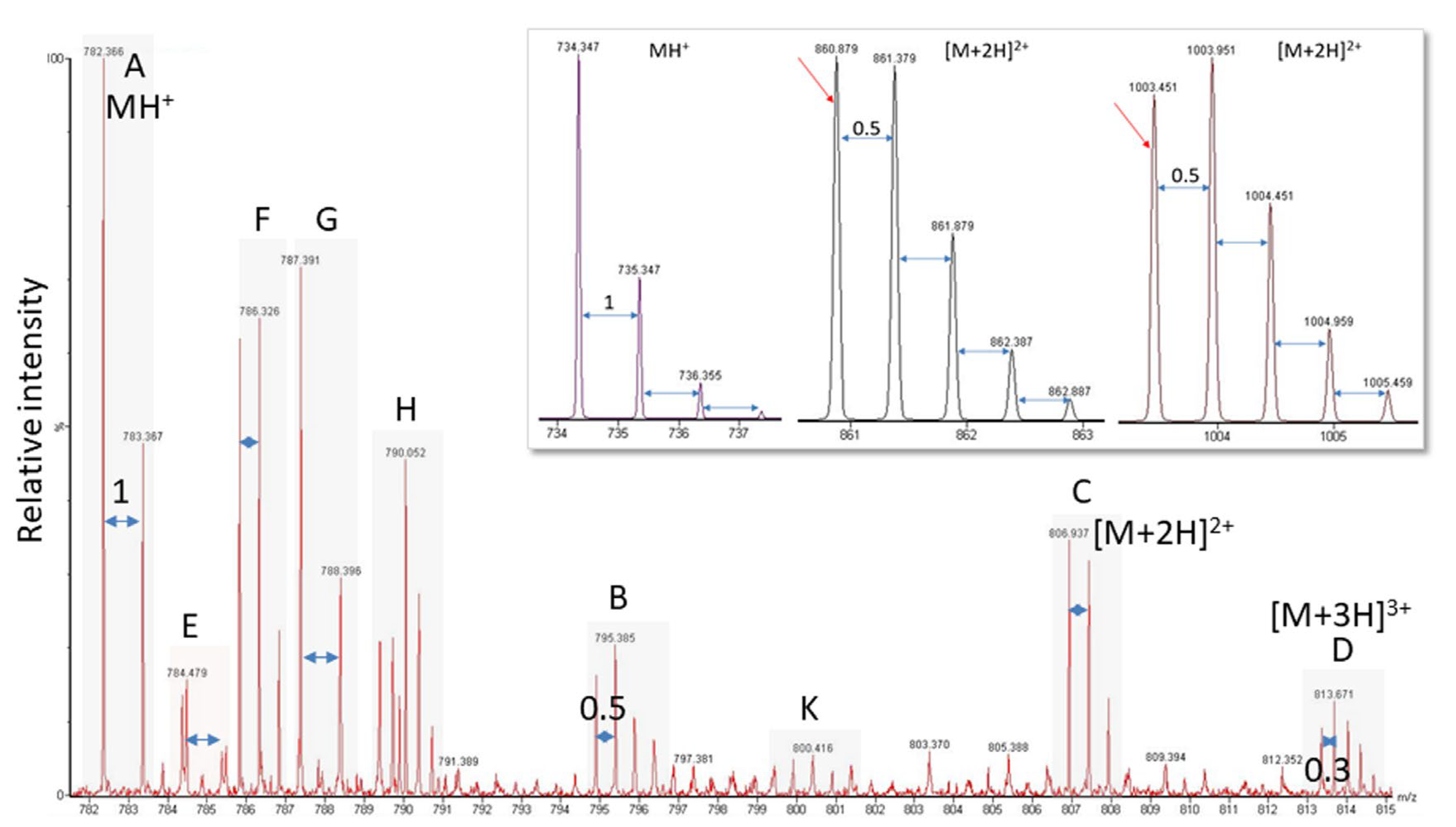
2.3. Low-Intensity Signals
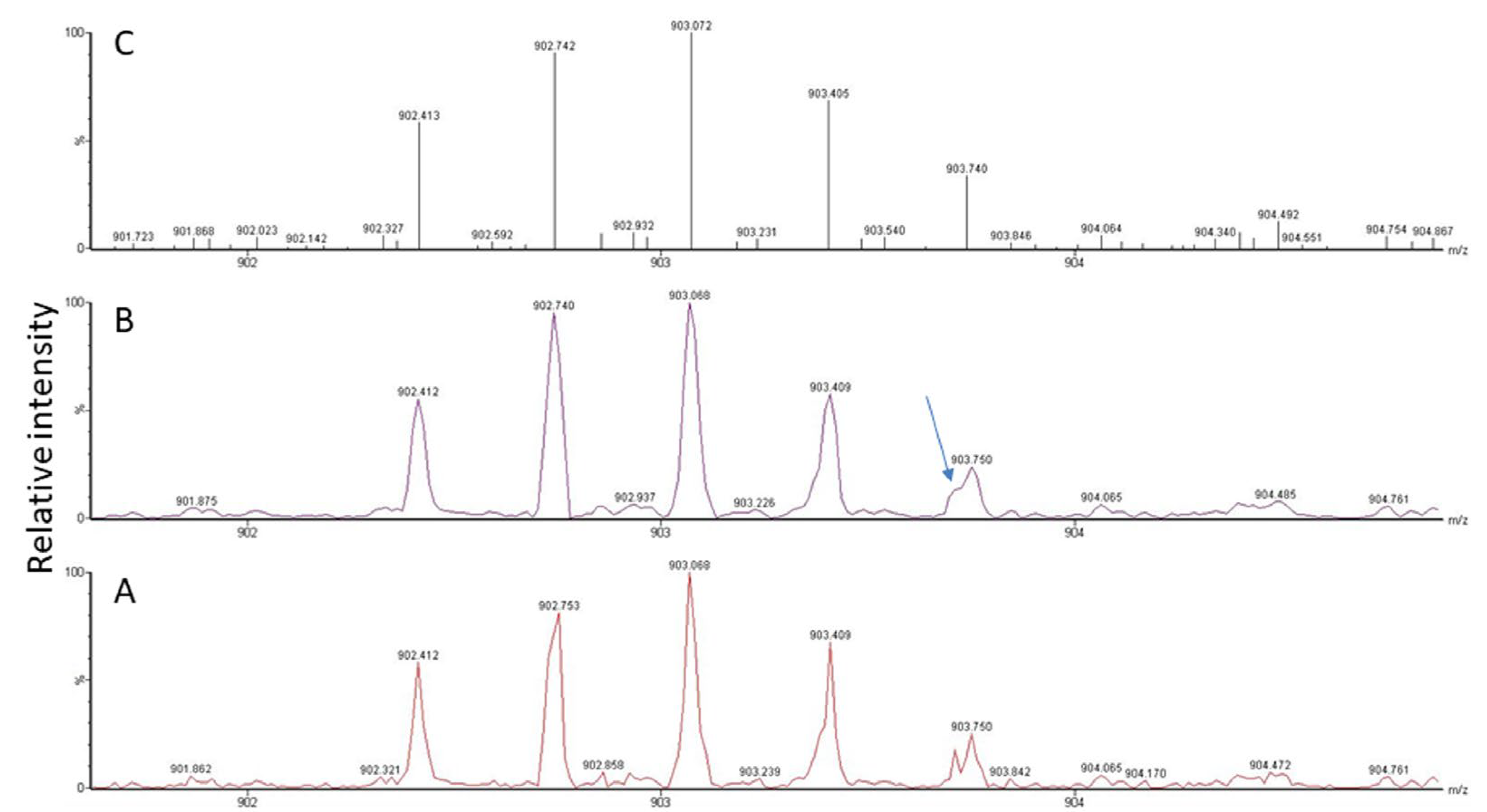
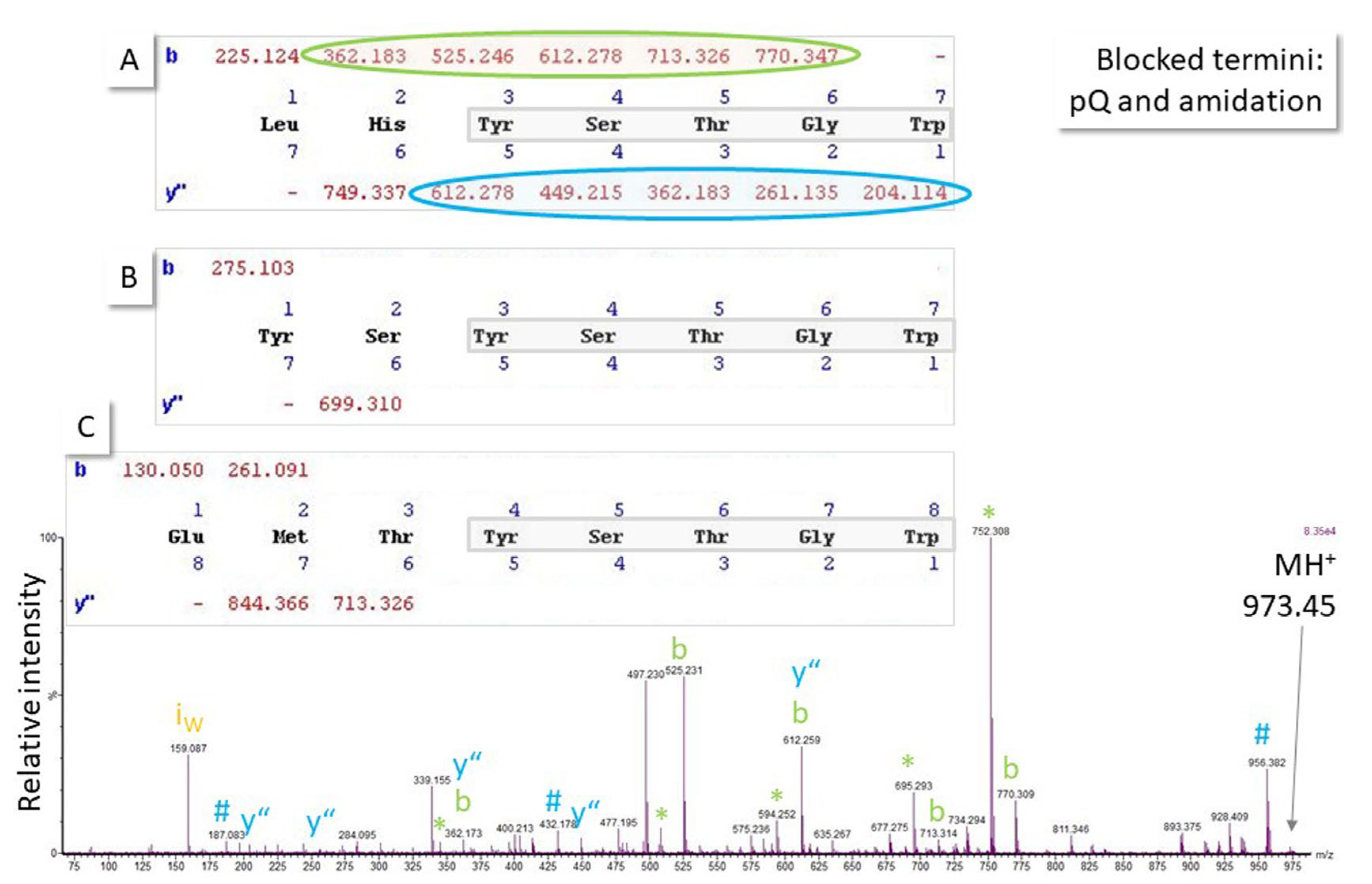
2.4. Ambiguous Spectra
3. Conclusions
4. Materials and Methods
4.1. Patients and Permissions
4.2. Protein Expression Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Nadler, W.M.; Waidelich, D.; Kerner, A.; Hanke, S.; Berg, R.; Trumpp, A.; Rösli, C. MALDI versus ESI: The impact of the ion source on peptide identification. J. Proteome Res. 2017, 16, 1207–1215. [Google Scholar] [CrossRef]
- Cui, M.; Cheng, C.; Zhang, L. High-throughput proteomics: A methodological mini-review. Lab. Investig. 2022, 102, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R., 3rd. Recent technical advances in proteomics. F1000Research 2019, 8, F1000 Faculty Rev-351–351. [Google Scholar] [CrossRef] [Green Version]
- Mann, M.; Kumar, C.; Zeng, W.F.; Strauss, M.T. Artificial intelligence for proteomics and biomarker discovery. Cell Syst. 2021, 12, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Mann, M. A beginner’s guide to mass spectrometry–based proteomics. Biochem 2020, 42, 64–69. [Google Scholar] [CrossRef]
- Schmidt, A.; Forne, I.; Imhof, A. Bioinformatic analysis of proteomics data. BMC Systems Biol. 2014, 8 (Suppl. 2), S3. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Hou, J.; Tanner, J.J.; Cheng, J. Bioinformatics methods for mass spectrometry-based proteomics data analysis. Int. J. Mol. Sci. 2020, 21, 2873. [Google Scholar] [CrossRef] [Green Version]
- Nakayasu, E.S.; Gritsenko, M.; Piehowski, P.D.; Gao, Y.; Orton, D.J.; Schepmoes, A.A.; Fillmore, T.L.; Frohnert, B.I.; Rewers, M.; Krischer, J.P.; et al. Tutorial: Best practices and considerations for mass-spectrometry-based protein biomarker discovery and validation. Nat. Protoc. 2021, 16, 3737–3760. [Google Scholar] [CrossRef] [PubMed]
- Coorssen, J.R.; Yergey, A.L. Proteomics is analytical chemistry: Fitness-for-purpose in the application of top-down and bottom-up analyses. Proteomes 2015, 3, 440–453. [Google Scholar] [CrossRef] [Green Version]
- König, S. Spectral quality overrides software score - A brief tutorial on the analysis of peptide fragmentation data for mass spectrometry laymen. J. Mass Spectrom. 2021, 56, e4616. [Google Scholar] [PubMed]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A critical review of bottom-up proteomics: The good, the bad, and the future of this field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef]
- Makarov, A.; Denisov, E.; Lange, O.; Horning, S. Dynamic range of mass accuracy in LTQ orbitrap hybrid mass spectrometer. J. Am. Soc. Mass Spectrom. 2006, 17, 977–982. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.; Page, J.S.; Smith, R.D. Charge competition and the linear dynamic range of detection in electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15, 1416–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, A.P.; Blakney, G.T.; Hendrickson, C.L.; Ellis, S.R.; Heeren, R.M.A.; Smith, D.F. Ultra-high mass resolving power, mass accuracy, and dynamic range MALDI mass spectrometry imaging by 21-T FT-ICR MS. Anal. Chem. 2020, 92, 3133–3142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, S.; Rasheed, F.; Zahra, R.; König, S. Gastric cancer pre-stage detection and early diagnosis of gastritis using serum protein signatures. Molecules 2022, 27, 2857. [Google Scholar] [CrossRef] [PubMed]
- Aziz, S.; Rasheed, F.; Zahra, R.; Akhter, T.S.; König, S. Microbial proteins in stomach biopsies associated with gastritis, ulcer, and gastric cancer. Molecules 2022, 27, 5410. [Google Scholar] [CrossRef]
- Wang, H.; Chen, X.L.; Liu, K.; Bai, D.; Zhang, W.H.; Chen, X.Z.; Hu, J.K. SIGES research group. Associations between gastric cancer risk and virus infection other than Epstein-Barr Virus: A systematic review and meta-analysis based on epidemiological studies. Clin. Transl. Gastroenterol. 2020, 11, e00201. [Google Scholar] [CrossRef]
- Takada, K. Epstein-Barr virus and gastric carcinoma. J. Clin. Pathol. Mol. Pathol. 2000, 53, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Firoz, A.; Ali, H.M.; Rehman, S.; Rather, I.A. Gastric cancer and viruses: A fine line between friend or foe. Vaccines 2022, 10, 600. [Google Scholar] [CrossRef]
- Ramamurthy, M.; Sankar, S.; Kannangai, R.; Nandagopal, B.; Sridharan, G. Application of viromics: A new approach to the understanding of viral infections in humans. Virusdisease 2017, 28, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Bikel, S.; Valdez-Lara, A.; Cornejo-Granados, F.; Rico, K.; Canizales-Quinteros, S.; Soberón, X.; Del Pozo-Yauner, L.; Ochoa-Leyva, A. Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: Towards a systems-level understanding of human microbiome. Comput. Struct. Biotechnol. J. 2015, 13, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Santiago-Rodriguez, T.M.; Hollister, E.B. Human virome and disease: High-throughput sequencing for virus discovery, identification of phage-bacteria dysbiosis and development of therapeutic approaches with emphasis on the human gut. Viruses 2019, 11, 656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillary, L.S.; Adriaenssens, E.M.; Jones, D.L.; McDonald, J.E. RNA-viromics reveals diverse communities of soil RNA viruses with the potential to affect grassland ecosystems across multiple trophic levels. ISME Commun. 2022, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Special Issue “Viromics: Approaches, advances and applications”. Viruses 2019. Available online: https://www.mdpi.com/journal/viruses/special_issues/viromics (accessed on 13 February 2023).
- Sender, R.; Bar-On, Y.M.; Gleizer, S.; Bernstein, B.; Flomholz, A.; Phillips, R.; Milo, R. The total number and mass of SARS-CoV-2 virions. Proc. Natl. Acad. Sci. USA 2021, 118, e2024815118. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aziz, S.; Rasheed, F.; Zahra, R.; König, S. Mass Spectrometry-Based Proteomics of Minor Species in the Bulk: Questions to Raise with Respect to the Untargeted Analysis of Viral Proteins in Human Tissue. Life 2023, 13, 544. https://doi.org/10.3390/life13020544
Aziz S, Rasheed F, Zahra R, König S. Mass Spectrometry-Based Proteomics of Minor Species in the Bulk: Questions to Raise with Respect to the Untargeted Analysis of Viral Proteins in Human Tissue. Life. 2023; 13(2):544. https://doi.org/10.3390/life13020544
Chicago/Turabian StyleAziz, Shahid, Faisal Rasheed, Rabaab Zahra, and Simone König. 2023. "Mass Spectrometry-Based Proteomics of Minor Species in the Bulk: Questions to Raise with Respect to the Untargeted Analysis of Viral Proteins in Human Tissue" Life 13, no. 2: 544. https://doi.org/10.3390/life13020544
APA StyleAziz, S., Rasheed, F., Zahra, R., & König, S. (2023). Mass Spectrometry-Based Proteomics of Minor Species in the Bulk: Questions to Raise with Respect to the Untargeted Analysis of Viral Proteins in Human Tissue. Life, 13(2), 544. https://doi.org/10.3390/life13020544