

The Role of Genetic Mutations in Mitochondrial-Driven Cancer Growth in Selected Tumors: Breast and Gynecological Malignancies


 ,
,
Abstract
:1. Introduction
2. Breast Cancer (BC)
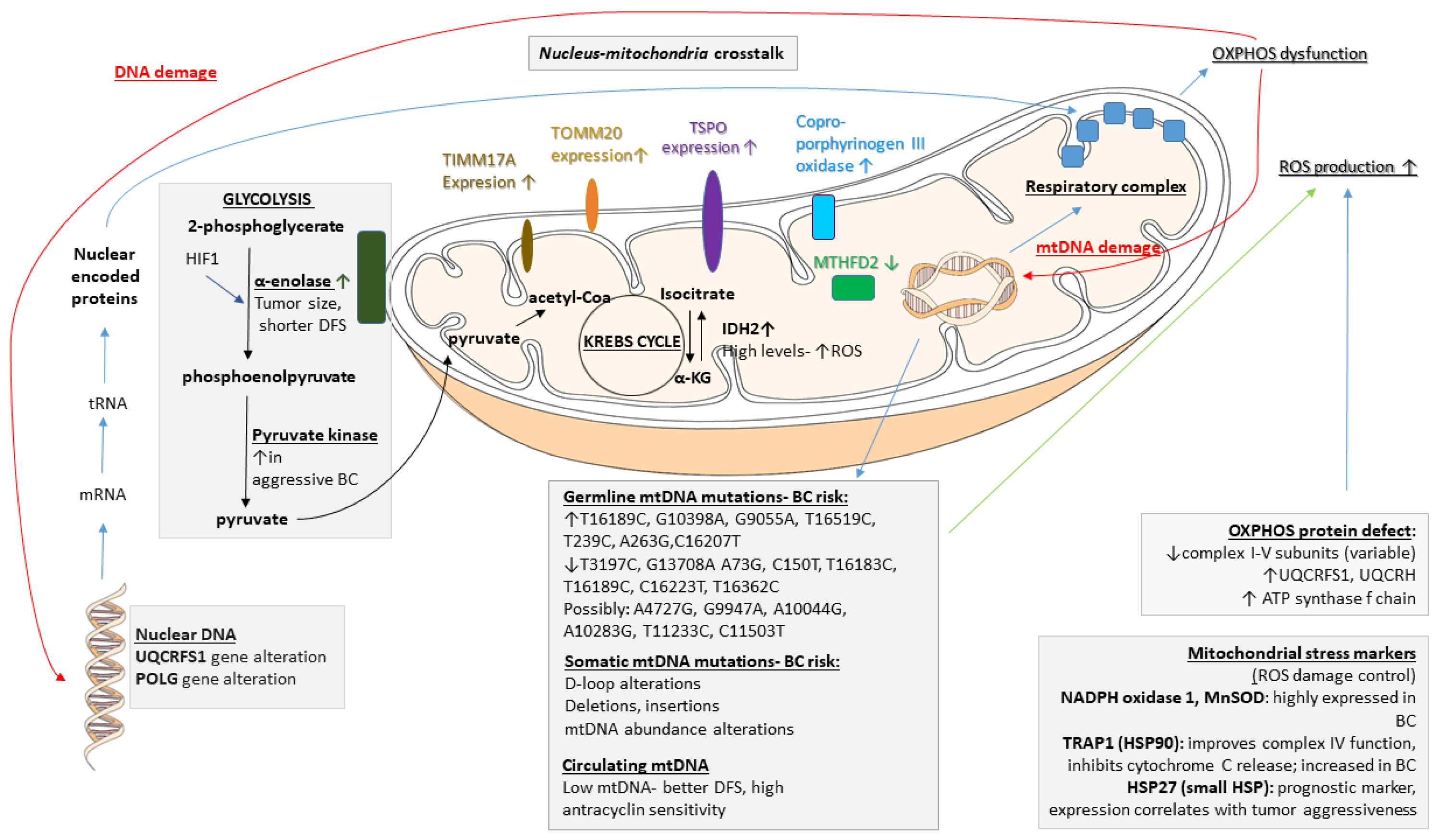
2.1. Mitochondria in BC Pathogenesis
- Enolases. Enolases catalyze the conversion of 2-phosphoglycerate to phosphoenolpyruvate. These enzymes are typically located within the cytosol, yet they tightly associate with the mitochondrial surface [24]. In human tissues, three genetic loci, namely, α, β, and γ, encode the different enolase isoforms. Enolase 1 is present in almost all adult tissues, enolase 2 is found in neuronal and neuroendocrine tissues, and enolase 3 is found mainly in muscle. The enzyme is upregulated under stress conditions via the activation of hypoxia-inducible factor-1 (HIF-1). The overexpression of α-enolase is associated with tumor development, which also serves as a potential diagnostic and prognostic marker [25]. In BC, α-enolase gene expression correlates with tumor size and a shorter disease-free interval [26].
- Pyruvate kinase. Pyruvate kinase (PK) is a rate-limiting glycolytic enzyme that converts phosphoenolpyruvate to pyruvate with the generation of one ATP molecule. It has two isoforms, PKM1 and PKM2, which are encoded by the same gene and are generated by alternative splicing. PKM1 is found mainly in normal cells, whereas PKM2 is an embryonic isoform that is expressed in cancer cells [27]. Elevated levels have been found to be associated with aggressive breast carcinomas [28].
- Hydratases. The NAD-dependent bifunctional methylenetetrahydrofolate dehydrogenase/cyclohydrolase (MTHFD) regulates the biosynthesis of tetrahydrofolate, providing precursors for nucleotides and methylation reactions. The MTHFD2 protein content is 3-fold decreased in BC lines [31].
- Dehydrogenases. Isocitrate dehydrogenases (IDHs) are important players in the exchange of metabolites within the cell, and two IDH isoforms can be found within the mitochondrion. IDH2, an NADP-dependent enzyme, has a role in the shuttling of electrons between the mitochondrion and the cytosol. IDH3 is an NAD-dependent mitochondrial matrix enzyme that is involved in the TCA cycle. BC cell lines display high levels of IDH2, and its expression is positively associated with overall survival in BC patients [32], possibly due to enhanced reactive oxygen species (ROS) protection.
- Oxidases. Coproporphyrinogen III oxidase (HemN), an enzyme required for heme synthesis, is present in the inner mitochondrial membrane. Its expression is increased in Adriamycin-resistant BC cells [33].
2.2. Mitochondrial DNA (mtDNA) and BC
- Germline mtDNA mutations. BC cells, like other cancer types, commonly harbor instability in the mitochondrial genome [68,69,70,71]. In this section, we discuss some of the widely investigated mtDNA polymorphisms that affect breast carcinogenesis. In the mtDNA T16189C germline mutation, various factors contribute to the substitution of T by C at nucleotide position (np) 16189, which is associated with susceptibility to BC development [72]. The 10398A allele of the NADH dehydrogenase-3 locus (ND3) of mtDNA is associated with an increased risk of invasive BC in African-American women [58,59] and in North Indian women [59]. The 10398G polymorphism of ND3 has been shown to increase the risk of BC in European American, Polish, and Malay populations [45,55,59,73,74]. It is also possible that polymorphisms in the mitochondrial genome could interact with life style and nutritional factors, such as alcohol consumption [75]. Chronic alcohol use may cause OXPHOS deficiency and other cellular changes. The mechanism by which the presence of these mutations leads to mitochondrial dysfunction is not clearly defined, but the G10398A variant of mtDNA may result in defective complex I function and thus lead to increased ROS production [59,76]. Whether ROS produced due to the G10398A polymorphism are sufficient to induce tumor formation remains to be determined, but the presence of other mutations combined with G10398A may contribute to breast tumorigenesis. Other single-nucleotide polymorphisms (SNPs) in mtDNA, including G9055A, T16519C, T239C, A263G, and C16207T, may also result in increased susceptibility to BC [45,73]. mtDNA T3197C and G13708A SNPs decrease the BC risk [73], and reduced incidences of mtDNA A73G, C150T, T16183C, T16189C, C16223T, and T16362C SNPs were noted in BC patients compared to database controls [46], along with other mtDNA polymorphisms associated with BC [77]. An analysis of the sequences of genes encoding complex I in cancer tissues and corresponding normal tissues led to the discovery of very rare mtDNA polymorphisms, including A4727G, G9947A, A10044G, A10283G, T11233C, and C11503T, that may have implications in BC development [46].
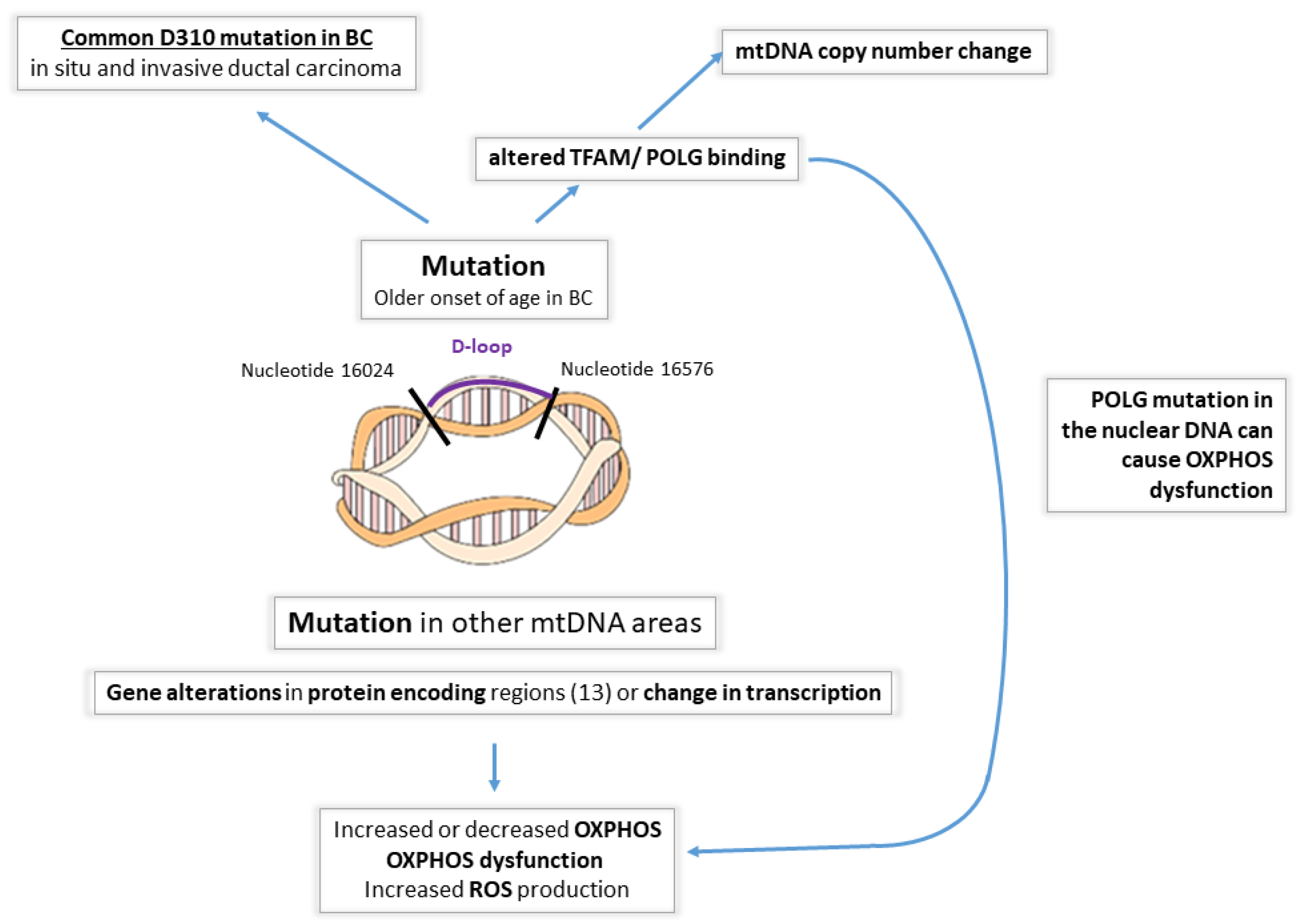
- Somatic mtDNA alterations. Despite the fact that numerous germline mutations have been linked to breast tumorigenesis, the majority of BCs are not inherited. In sporadic BC cases, somatic mtDNA mutations may lead to the selective transformation of breast epithelial cells and tumorigenesis. Various somatic mtDNA mutations have been detected in BC [39,42,50,61,78,79,80,81,82,83,84,85,86]. The majority of somatic mtDNA mutations occur in the D-loop region and can be point mutations, deletions, insertions, or missense mutations.
- mtDNA displacement loop alterations (Figure 2). The D-loop is considered a hot spot for mutations [79] and is up to ~60 times more susceptible to mutations than the coding regions, according to some studies. The increase in susceptibility, however, is variable among different studies, with some showing only a 7-fold increase [60]. The D-loop itself is a noncoding region, but mutations in this area are typically significant and potentially affect the expression of mtDNA-encoded protein/s or alter mtDNA replication. The replication of mtDNA starts in the displacement loop (D-loop) region located between nucleotides 16024 and 16576. mtDNA replication involves DNA polymerase γ (POLG) and mitochondrial transcription factor A (TFAM), the latter being the key transcription factor regulating mtDNA copy numbers [87,88]. In BC patients, the occurrence of D-loop mutations is associated with an older age of onset [61]. A homopolymeric C-stretch within the D-loop, termed the 310 microsatellite sequence, is a relatively conserved region that includes the replication origin of the mtDNA heavy strand [89]. Previous reports have shown D310 sequence alterations in human cancers, including ductal in situ carcinomas (68%) and invasive ductal carcinomas (71%) [57]. In another small study, 11 of 18 BCs harbored mtDNA mutations, of which 42% were D310 alterations [39]. Histologically normal breast epithelial cells adjacent to invasive ductal carcinomas that carry D310 mutations may already represent tumor cell clonal expansion [57]. However, these may not be representative of a larger cohort.
- Deletions. Deletion of 4977 base pairs (ΔmtDNA4977 mutation) has been found in BC tissue, but it was also detected in the surrounding normal breast tissue—indicating either the premalignant state of the tissue exhibiting normal morphology, or representing a clinically non-significant alteration [90,91]. In addition, another research reported conflicting data on the role of ΔmtDNA4977 mutation in BC [61]. Later studies, however, demonstrated that the ΔmtDNA4977 mtDNA deletion, when associated with significant other nuclear gene alterations, such as in the BRCA, ER or TP53 genes, led to premature aging and breast tumorigenesis [92,93].
- 5.
- Alterations in mtDNA abundance. Mitochondria have multiple copies of mtDNA, and this copy number changes in response to energy demands, with both increased and decreased mtDNA content previously reported in cancer cells [94,95]. In the majority of BCs, the mtDNA content was decreased compared to the adjacent histologically normal tissue when measuring the mean mtDNA content using quantitative RT-PCR and ND1 gene primers [61].
2.3. Nuclear DNA Alterations Affecting Mitochondrial Function in Cancer
2.4. Mitochondrial Stress Markers in BC
- ROS damage control. NADPH oxidase 1, a major source of ROS in cells, predominantly localizes to the mitochondria and is highly expressed in breast (86%) tumors [102]. To counteract the damaging effects of ROS, cells contain a multilayered system of antioxidant defenses executed by three types of enzymes: superoxide dismutases (SODs), peroxidases (PODs), and catalases (CATs). MnSOD is constitutively present in the mitochondrial matrix, but its expression can be further induced by hypoxia. In BC patients, strong MnSOD staining can be observed in neoplastic cells, with moderate-to-strong staining in adjacent hyperplastic ducts and weak-to-moderate staining in the normal epithelium [106]. A histochemical study shows lower expression in BC cells compared to the adjacent normal epithelia [107].
- HSP90 family. Members of the HSP90 gene family are considered essential regulators of protein folding. TNF receptor-associated protein 1 (TRAP1) is a member of the HSP90 family and is considered mostly mitochondrial. In vivo studies in rats have shown that TRAP1 protects against hypoxia by reducing the generation of ROS, improving mitochondrial complex IV activity, and preserving ATP levels [108]. TRAP1 expression is induced in tumor cells. As shown by immunohistochemistry (IHC), TRAP1 staining appears intense in breast adenocarcinomas, while the normal matched epithelia stain weakly [109]. There is also evidence pointing to the anti-apoptotic role of the HSP90 family. TRAP1 and HSP90 are involved in the mitochondrial pathway that antagonizes the proapoptotic activity of cyclophilin D [109]. This interaction occurs in a multichaperone complex that is selectively assembled in tumor cells and is not present in normal mitochondria [110]. TRAP1 has also been shown to directly interact with members of the MPTP, inhibiting its opening and the subsequent release of cytochrome c (CytC) [111].
- Small HSP family. HSP27 is mainly cytosolic, but a small fraction localizes to the mitochondria. HSP27 expression may function as a useful prognostic marker of poor survival in many human cancers. HSP27 is upregulated in the serum of BC patients [112] and correlates with poor clinical outcomes. A clinical evaluation of BC patients showed the correlated expression of HSP27 with tumor aggressiveness and decreased survival [113].
2.5. Mitochondrial Membrane Markers and BC
2.6. Genetic Background and Mitochondria in BC
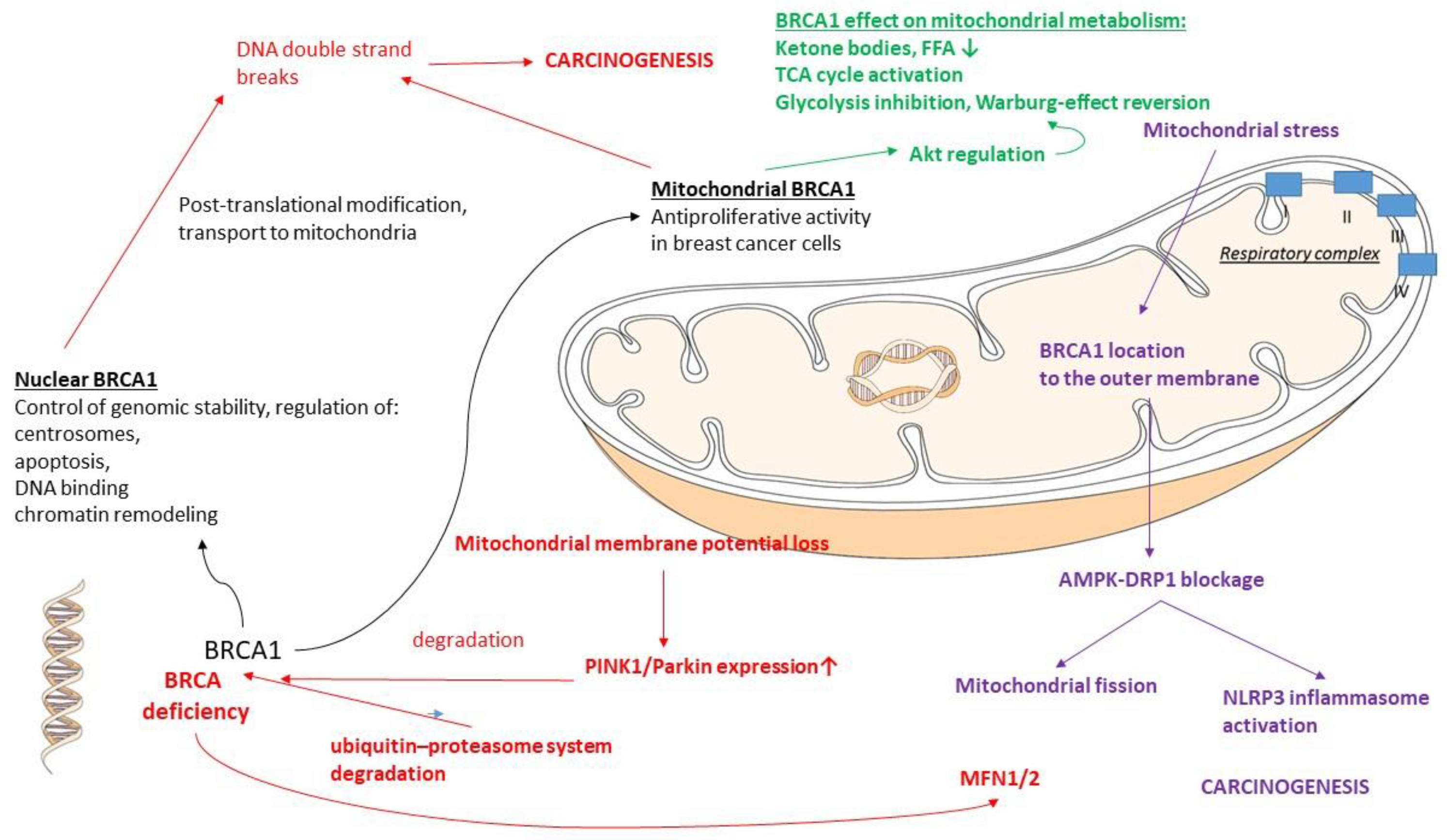
2.6.1. BRCA1
2.6.2. BRCA2
2.6.3. ERBB2 (HER2/Neu)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Driver Gene | Effects on Mitochondrial Metabolism |
|---|---|
| BRCA1 | Warburg effect reversal (glycolysis inhibition) [179] Activation of TCA cycle [179] Activation of OXPHOS [179] Mitochondrial BRCA1: antiproliferative activity [158] |
| BRCA2 | Mutation causes elevation of intracellular ROS production; oxidative stress causes mitochondrial dysfunction [180] |
| ErbB2 (HER2/Neu) | Promotes cancer cell growth and glycolysis [105,189] Mitochondrial ErbB2: enhances cellular glycolysis [189] |
2.6.4. PTEN
3. Endometrial Cancer (EC)
- Ultramutated (polymerase ε (POLE) mutant): mostly composed of endometrioid ECs, which, despite having an increased mutation frequency and hotspot mutations in the POLE gene (encoding the central catalytic subunit of DNA polymerase epsilon), have a better prognosis than other groups.
- Hypermutated (mismatch-repair-deficient (MMRd)): Involves germline and somatic mutations, resulting in microsatellite instability, such as via MLH1 silencing due to hypermethylation. In general, tumors in this group are associated with intermediate, stage-dependent prognosis.
- Copy number high (p53-abnormal): TP53 alterations are present, with ~50% of cases being serous carcinomas and carcinosarcomas, and ~25% of cases are higher-grade endometrioid ECs. In general, tumors in this group are associated with inferior survival.
- Copy number low (no specific molecular profile (NSMP)): This group includes TP53 and POLE wild-type and MMR-proficient tumors, which frequently harbor PTEN, PIK3CA, ARID1A, or KRAS alterations. The majority of these tumors are low-grade endometrioid ECs [191].
3.1. EC and Mitochondrial Changes
3.2. PTEN
3.3. PIK3CA
3.4. KRAS
3.5. CTNNB1
- “Wnt ON”: Wnt binds to its membrane receptor (the so-called Fz and LRP5/6 receptors) → this induces the cytoplasmic disheveled (DVL) protein, recruited by the Fz receptor → cytoplasmic LRP5/6 protein phosphorylation and Axin protein recruitment → no β-catenin phosphorylation by Axin → no β-catenin degradation → β-catenin accumulates in the nucleus and displaces Groucho/TLE from the TCF-TLE complex, allowing TCF to activate Wnt-responsive genes.
- “Wnt OFF”: Absent Wnt → no receptor binding and activation → cytoplasmic β-catenin forms a complex with Axin, GSK3, and CK1→ β-catenin phosphorylation by Axin and GSK3 → E3 ubiquitin ligase β-Trcp recognizes phosphorylated β-catenin → β-catenin proteasomal degradation → TCF-TLE complex and histone deacetylases (HDACs) repress Wnt target genes [269,270,271].
3.6. FGF/FGFR Pathway
3.7. TP53
4. Epithelial Ovarian Carcinomas (OCs)
4.1. OC and Mitochondria
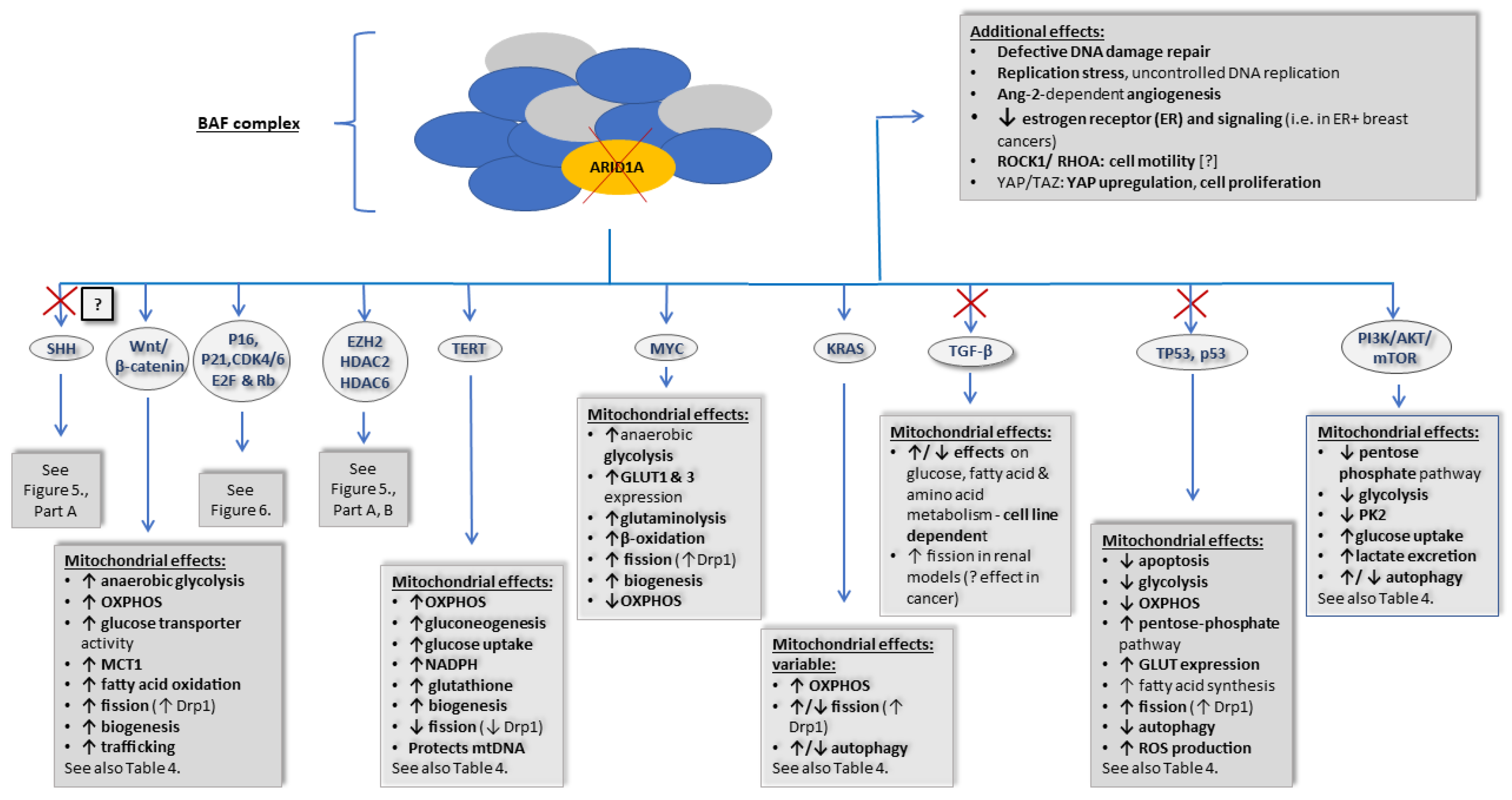
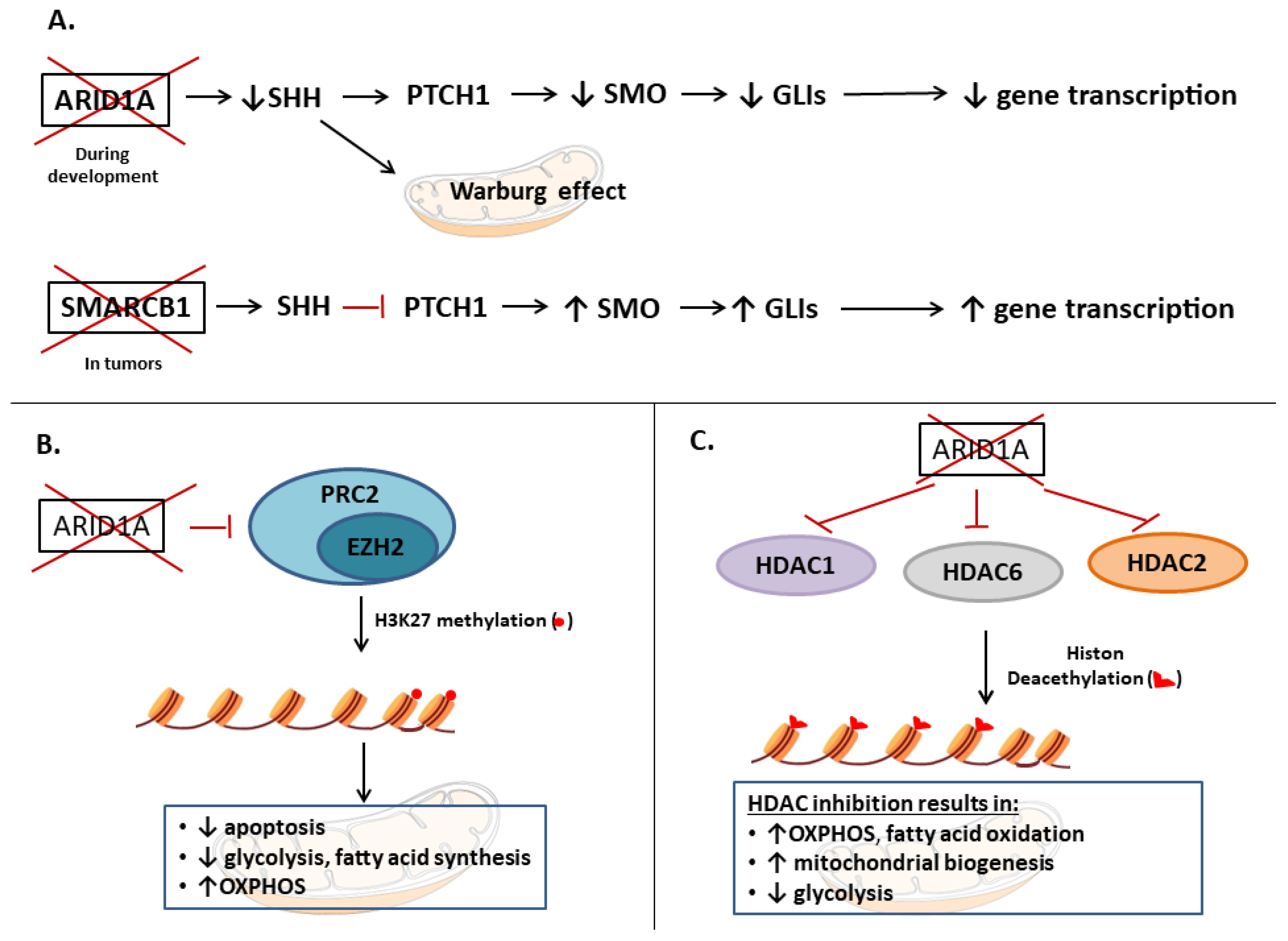
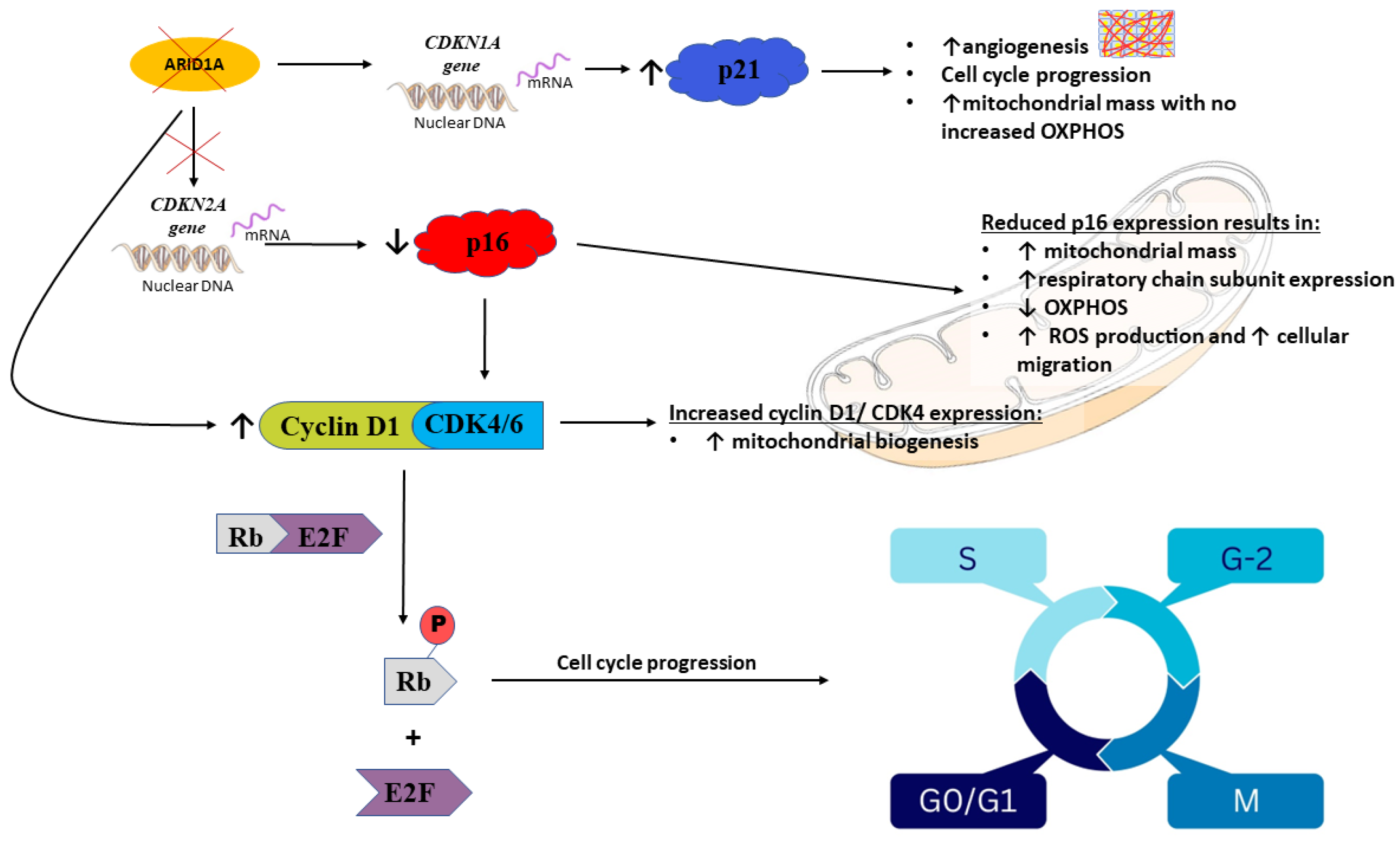
4.2. ARID1A
- EZH2, histone modifications (HDACs);
- SHH (ARID1A loss, possibly resulting in inhibition);
- p16 and p21, CDK4/5, Rb, and E2F4 (ARID1A loss, resulting in inhibition);
- Wnt/β-catenin (ARID1A loss, resulting in activation);
- TP53, p53 (ARID1A loss, resulting in inhibition);
- TERT (ARID1A loss, resulting in activation);
- Transforming growth factor β (TGFβ) (ARID1A loss, resulting in inhibition);
- MYC (ARID1A loss, resulting in activation);
- KRAS (ARID1A loss, resulting in activation);
- PI3K/AKT/mTOR (ARID1A loss, resulting in activation).
4.2.1. EZH2 and HDACs
4.2.2. SHH Pathway
4.2.3. p16 and p21, CDK4/5, Rb, and E2F4
4.2.4. Wnt/β-Catenin Pathway, TP53/p53, TERT, and KRAS
4.2.5. Transforming Growth Factor β (TGF-β)
4.2.6. MYC
4.2.7. PI3K/AKT/mTOR
4.3. TERT
| Gene/Protein Name Gene Effect | General Effects ± Main Downstream Pathways | Effect on Mitochondria |
|---|---|---|
| PTEN /PTEN Tumor suppressor | PI3K/AKT/mTOR (activated via PTEN loss) and non-enzymatic roles PTEN loss results in: ↑ cell proliferation ↑ cell growth ↑ cell survival, migration, cell adhesion ↑ angiogenesis | PTEN: - ↑ apoptosis induction [226] - ↑ ROS production [226] Contradictory data on autophagy: - ↑ autophagy and lysosomal mass [227,228,229,230] or - ┤mitophagy via blocking the TLR4–JNK–BNIP3 pathway [231,232] -┤mitophagy via ubiquitin dephosphorylation [231,232] - ┤mitophagy via ↑ Mfn2 and ↓Rab7a [231,232] Loss of PTEN: - ↑ glycolysis [233] - ┤ gluconeogenesis [233] - ↑ lipogenesis [233] - ↑ mitochondrial biogenesis [233] |
| PIK3CA /PI3K Proto-oncogene | PI3K-AKT-mTOR pathway PIK3CA activation results in: ↑ cell growth ↑ motility ↑ survival and proliferation ↑ protein synthesis ↑ intracellular trafficking ↑ angiogenesis | PI3K/AKT/mTOR pathway: ┤ pentose phosphate pathway (via G6PD stabilization) [250] ┤PK2, a rate-limiting enzyme of glycolysis [250] ↑ glucose uptake [250] ↑ lactate excretion [250] ┤autophagy (at moderate ROS levels) [254] ↑ autophagy (at moderate ROS levels) [254] ┤PI3K-AKT-mTOR pathway: - ↓ intracellular lipid accumulation via ↓ de novo fatty acid synthesis [251], ↓ FASN [252], ↓ SREBP [252], and ↑ fatty acid oxidation [251] Contradictory data on mitochondrial trafficking: - PI3K and mTOR inhibitors: ┤ tubulin polymerization, leading to microtubule disturbance [256] or - PI3K inhibitor: ↑ mitochondrial trafficking [257] |
| KRAS/KRAS Proto-oncogene | Major downstream pathways: PI3K, MAPK, and Ral small GTPase KRAS activation results in: - ↑ proliferation, transformation - Cell survival | - ↑ mitochondrial fission (↑ Drp1) [1,262,263,264] - ↑ mitophagy [1,262,263,264] - ↑ OXPHOS [1,262,263,264] The effect of tumor-suppressive therapy in RAS-driven tumors: - ┤Drp1 [262,263,264] - ↑ Mfn2 expression (mitochondrial fusion induced by doxycycline/leflunomide) [263] - ↓ autophagy proteins [265] |
| CTNNB1 /β-catenin Proto-oncogene | Major downstream pathways: β-Catenin regulates the expression of many Wnt target genes, including MYC, CCND1, and CDKN1A General effects: - ↑ proliferation - regulation of cellular development and differentiation - ↑ angiogenesis - regulation of migration and invasion - regulation of cellular homeostasis | Wnt/β-catenin activation: - ↑ anaerobic glycolysis (↑ PDK1, ↑LDH-A) [272,273,274,275,276,277,278] - ↑ OXPHOS (typically less increment than anaerobic glycolysis) [275,276,277,278,279] - ↑ glucose transporter activity [277] - ↑ MCT1 [272,278] - ↑ fatty acid oxidation [280] - ↑ mitochondrial fission (↑ Drp1) [282] - ↑ apoptosis (although under special circumstances, the opposite is true) [283] Wnt/β-catenin signaling inhibition: ↓ anaerobic glycolysis (↓ PDK1) [273] ↓ SREBP-1c in hepatocytes [281] |
| FGFRs (1–4) /FGFRs (1–4) Proto-oncogenes | Major downstream pathways: PI3K/AKT/mTOR, RAS/RAF/MEK/ERK1/2 or MAPK, PIP2/DAG/PKC, STAT, p53, and β-catenin pathways - Development - Cell proliferation - Apoptosis regulation - Cell migration - Angiogenesis | FGF19: [308] ↓ gluconeogenesis ↑ glycogen synthesis ↑ peripheral insulin sensitivity ↑ glucose metabolism ↓ lipogenesis ↑ fatty acid oxidation FGF21: [309,310] - ↑ PGC-1α - ↑ mitochondrial ATP production - ↑ hepatic gluconeogenesis - ↑ ketogenesis Mitochondrial FGFR1-like receptor [310]: - ↑ PDHK1 ┤PDH → ↓ pyruvate to acetyl-CoA conversion → ↓ glycolysis α/βKlotho (tumor suppressor effects—some effects only seen in tumor cells) [311,312,313]: ↓ glycolysis (via HK, PFK-1, PK2, PDHK1) ↓ fatty acid synthesis ↓ GLUT expression (GLUT1, GLUT4) ↓ lactate transporter expression (MCT4) ↓ ROS production ┤autophagy |
| TP53 /p53 Tumor suppressor | Wild-type TP53: - Cell cycle arrest - Growth arrest - DNA repair - ↑ senescence | Wild-type TP53: [1] - ↑ apoptosis (↓ Bcl2 and ↓ BclXl; ↑ Bax and ↑ Bak), - ↑ glycolysis - ↑ OXPHOS - ┤ pentose phosphate pathway - ↓ glucose receptor expression (GLUT1, GLUT3, GLUT4) - ┤ fatty acid synthesis - ┤ mitochondrial fission (via Drp1) - ↑ autophagy (via mTOR-dependent and independent manner) - ↓ ROS production |
| ARID1A Tumor suppressor | - Cellular differentiation - Cell cycle regulation - Cell migration - Angiogenesis - DNA repair For downstream pathways, see Figure 4, Figure 5 and Figure 6 | ARID1A loss: - ↑ mitochondrial membrane potential [391] - ↑ OXPHOS [2,391,392,393] - ↑ anaerobic glycolysis [393] - ↑ mitochondrial mass [391] - ↑ mitochondrial fission [391] For details on its downstream pathways, see Figure 4, Figure 5 and Figure 6 |
| TERT Proto-oncogene | Canonical function: - Telomere elongation Noncanonical functions: - Chromatin structure regulation - RNA silencing - Epigenetic changes - Mitochondrial effects - Activation of signaling pathways (i.e., NF-κB and Wnt/β-catenin signaling pathways) - ↑ cell adhesion and migration | TERT expression/overexpression: - ↑ or ↓ apoptosis [430] - Directly binds to mtDNA and protects it from ROS-induced damage [433] - ↑ expression of glycolysis enzymes [434] - ↑ glucose flux via the pentose phosphate pathway, ↑ NADPH [435] - ↑ glutathione levels [436] - ↑ mitochondrial mass [434] Loss of mitochondrial TERT: - ↑ autophagy [436] Telomere dysfunction: - ↓ PGC-1α and PGC-1β promoters (decreasing mitochondrial biogenesis) [432] |
| FGF Subfamily | FGFs | Additional Information | FGF Associations with BC, EC, or OC (Presence of Immunoreactivity (IR) [291] or Increased FGF Gene Expression/Activating Mutation/Gene Amplification [291,292]) |
|---|---|---|---|
| 1 | 1, 2 | “Paracrine” FGFs; Bind to FGFRs via HSPG | 1: OC (gene amplification) |
| 4 | 4, 5, 6 | “Paracrine” FGFs; Bind to FGFRs via HSPG | 4: BC, EC (both: rare, IR; BC: + gene amplification) |
| 7 | 3, 7, 10, 22 | “Paracrine” FGFs; Bind to FGFRs via HSPG | 3: BC (rare: IR; + gene amplification) 7: EC (rare, weak IR) 10: BC (gene overexpression) |
| 8 | 8, 17, 18 | “Paracrine” FGFs; Bind to FGFRs via HSPG | 8: EC (increased RNA expression) 17: BC, OC (both weak staining, OC: rare) 18: BC (rare) |
| 9 | 9, 16, 20 | “Paracrine” FGFs; Bind to FGFRs via HSPG | 9: EC, OC (both: IR; EC: + gene mutation) 16: OC (gene overexpression) 20: EC (increased RNA expression) |
| 19 | 19, 21, 23 | “Endocrine” FGFs; Bind to FGFRs via α/or βKlotho proteins (obligatory co-receptors) | 19: OC, EC (EC: rare and weak staining) αKlotho: tumor suppressor in BC, and OC [293,294] βKlotho: tumor suppressor in EC |
| 11 | 11, 12, 13, 14 | “Paracrine” FGFs; Bind to FGFRs via HSPG; intracellular localization and binding is typical [295,296,297] | - |
| Function | Subunits/Alternative Names |
|---|---|
| Catalytic ATP-ases | SMARCA2/BRM SMARCA4/BAF250B/BRG1 |
| Core subunits | SMARCB1/SNF5/INI1 SMARCC1/BAF155 SMARCC2/BAF170 |
| Signature subunits (BAF) | ARID1A/SMARCCF1/BAF250A ARID1B/BAF250B |
| Signature subunits (PBAF) | ARID2/BAF200 |
| Accessory subunits | ACTL6A, or B/BAF53A, or B SMARCD1, 2, or 3/BAF60A, B, or C SMARCE1/BAF57 DPF1,2, or 3/BAF45B, C, or D PHF10/BAF45A BRD7, or 9 BCL11A, or B BCL7A, B, or C SS18 |
5. Similarities and Differences between BCs, ECs, and OCs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Czegle, I.; Gray, A.L.; Wang, M.; Liu, Y.; Wang, J.; Wappler-Guzzetta, E.A. Mitochondria and Their Relationship with Common Genetic Abnormalities in Hematologic Malignancies. Life 2021, 11, 1351. [Google Scholar] [CrossRef] [PubMed]
- Emmings, E.; Mullany, S.; Chang, Z.; Landen, J.C.N.; Linder, S.; Bazzaro, M. Targeting Mitochondria for Treatment of Chemoresistant Ovarian Cancer. Int. J. Mol. Sci. 2019, 20, 229. [Google Scholar] [CrossRef] [Green Version]
- Czarnecka, A.M.; Klemba, A.; Semczuk, A.; Plak, K.; Marzec, B.; Krawczyk, T.; Kofler, B.; Golik, P.; Bartnik, E. Common mitochondrial polymorphisms as risk factor for endometrial cancer. Int. Arch. Med. 2009, 2, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, G.R.; Wardell, S.E.; Cakir, M.; Yip, C.; Ahn, Y.R.; Ali, M.; Yllanes, A.P.; Chao, C.A.; McDonnell, D.P.; Wood, K.C. Dysregulation of mitochondrial dynamics proteins are a targetable feature of human tumors. Nat. Commun. 2018, 9, 1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, P.C.; Herrmann, E.C. Oxygen metabolism and a potential role for cytochrome c oxidase in the Warburg effect. J. Bioenerg. Biomembr. 2007, 39, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Cormio, A.; Cormio, G.; Musicco, C.; Sardanelli, A.M.; Gasparre, G.; Gadaleta, M.N. Mitochondrial changes in endometrial carcinoma: Possible role in tumor diagnosis and prognosis (Review). Oncol. Rep. 2014, 33, 1011–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, P.; Singh, K.K. The mitochondrial landscape of ovarian cancer: Emerging insights. Carcinogenesis 2021, 42, 663–671. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, W.; Idowu, M.O.; Oh, U.; Wang, X.-Y.; Temkin, S.M.; Fang, X. Ovarian Cancer Relies on Glucose Transporter 1 to Fuel Glycolysis and Growth: Anti-Tumor Activity of BAY-876. Cancers 2018, 11, 33. [Google Scholar] [CrossRef] [Green Version]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-L.; Gong, Y.; Ji, P.; Xie, Y.-F.; Jiang, Y.-Z.; Liu, G.-Y. Targeting nucleotide metabolism: A promising approach to enhance cancer immunotherapy. J. Hematol. Oncol. J. Hematol. Oncol. 2022, 15, 45. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McTiernan, A. Behavioral risk factors in breast cancer: Can risk be modified? Oncologist 2003, 8, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, S.; Ellis, I.; Schnitt, S. WHO Classification of Tumours of the Breast, WHO Classification of Tumours, 4th ed.; IARC Press: Geneva, Switzerland, 2020; Volume 4. [Google Scholar]
- do Nascimento, R.G.; Otoni, K.M. Histological and molecular classification of breast cancer: What do we know? Mastology 2020, 30, e20200024. [Google Scholar] [CrossRef]
- Hammond, M.E.H. ASCO-CAP guidelines for breast predictive factor testing: An update. Appl. Immunohistochem. Mol. Morphol. AIMM 2011, 19, 499–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, A.C.; Hammond, M.E.H.; Allison, K.H.; Harvey, B.E.; Mangu, P.B.; Bartlett, J.M.S.; Bilous, M.; Ellis, I.O.; Fitzgibbons, P.; Hanna, W.; et al. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 2105–2122. [Google Scholar] [CrossRef] [Green Version]
- Duffy, M.J.; Harbeck, N.; Nap, M.; Molina, R.; Nicolini, A.; Senkus, E.; Cardoso, F. Clinical use of biomarkers in breast cancer: Updated guidelines from the European Group on Tumor Markers (EGTM). Eur. J. Cancer Oxf. Engl. 1990 2017, 75, 284–298. [Google Scholar] [CrossRef] [Green Version]
- Penault-Llorca, F.; Radosevic-Robin, N. Ki67 assessment in breast cancer: An update. Pathology 2017, 49, 166–171. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.-J. Panel members Personalizing the treatment of women with early breast cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef]
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E.; ESMO Guidelines Committee. Electronic address: [email protected] Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1194–1220. [Google Scholar] [CrossRef] [Green Version]
- De Paepe, B. Mitochondrial Markers for Cancer: Relevance to Diagnosis, Therapy, and Prognosis and General Understanding of Malignant Disease Mechanisms. ISRN Pathol. 2012, 2012, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Altenberg, B.; Greulich, K.O. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 2004, 84, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Entelis, N.; Brandina, I.; Kamenski, P.; Krasheninnikov, I.A.; Martin, R.P.; Tarassov, I. A glycolytic enzyme, enolase, is recruited as a cofactor of tRNA targeting toward mitochondria in Saccharomyces cerevisiae. Genes Dev. 2006, 20, 1609–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capello, M.; Ferri-Borgogno, S.; Cappello, P.; Novelli, F. α-Enolase: A promising therapeutic and diagnostic tumor target. FEBS J. 2011, 278, 1064–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, S.-H.; Chang, C.-C.; Chen, C.-S.; Tam, K.-W.; Wang, Y.-J.; Lee, C.-H.; Lin, H.-W.; Cheng, T.-C.; Huang, C.-S.; Chu, J.-S.; et al. Increased expression of enolase alpha in human breast cancer confers tamoxifen resistance in human breast cancer cells. Breast Cancer Res. Treat. 2010, 121, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef]
- Lüftner, D.; Mesterharm, J.; Akrivakis, C.; Geppert, R.; Petrides, P.E.; Wernecke, K.D.; Possinger, K. Tumor type M2 pyruvate kinase expression in advanced breast cancer. Anticancer Res. 2000, 20, 5077–5082. [Google Scholar]
- Owens, K.M.; Kulawiec, M.; Desouki, M.M.; Vanniarajan, A.; Singh, K.K. Impaired OXPHOS complex III in breast cancer. PLoS ONE 2011, 6, e23846. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, Y.; Kaneko, S.J.; Cupples, T.E.; Young, S.R. Ubiquinol cytochrome c reductase (UQCRFS1) gene amplification in primary breast cancer core biopsy samples. Gynecol. Oncol. 2004, 93, 54–58. [Google Scholar] [CrossRef]
- Yu, K.-D.; Chen, A.-X.; Yang, C.; Qiu, L.-X.; Fan, L.; Xu, W.-H.; Shao, Z.-M. Current evidence on the relationship between polymorphisms in the COX-2 gene and breast cancer risk: A meta-analysis. Breast Cancer Res. Treat. 2010, 122, 251–257. [Google Scholar] [CrossRef]
- Geiger, T.; Madden, S.F.; Gallagher, W.M.; Cox, J.; Mann, M. Proteomic portrait of human breast cancer progression identifies novel prognostic markers. Cancer Res. 2012, 72, 2428–2439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strong, R.; Nakanishi, T.; Ross, D.; Fenselau, C. Alterations in the mitochondrial proteome of adriamycin resistant MCF-7 breast cancer cells. J. Proteome Res. 2006, 5, 2389–2395. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.L.; Bohr, V.A. Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells. J. Biol. Chem. 1997, 272, 25409–25412. [Google Scholar] [CrossRef] [Green Version]
- Verma, M.; Naviaux, R.K.; Tanaka, M.; Kumar, D.; Franceschi, C.; Singh, K.K. Meeting report: Mitochondrial DNA and cancer epidemiology. Cancer Res. 2007, 67, 437–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, N.; Chandra, D. Mitochondrial DNA mutations and breast tumorigenesis. Biochim. Biophys. Acta 2013, 1836, 336–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.K.; Ayyasamy, V.; Owens, K.M.; Koul, M.S.; Vujcic, M. Mutations in mitochondrial DNA polymerase-gamma promote breast tumorigenesis. J. Hum. Genet. 2009, 54, 516–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, D.K.; Green, P.D.; Santos, J.H.; D’Souza, A.D.; Walther, Z.; Martin, W.D.; Christian, B.E.; Chandel, N.S.; Shadel, G.S. Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC(Min/+) mice. Am. J. Pathol. 2012, 180, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Parrella, P.; Xiao, Y.; Fliss, M.; Sanchez-Cespedes, M.; Mazzarelli, P.; Rinaldi, M.; Nicol, T.; Gabrielson, E.; Cuomo, C.; Cohen, D.; et al. Detection of mitochondrial DNA mutations in primary breast cancer and fine-needle aspirates. Cancer Res. 2001, 61, 7623–7626. [Google Scholar]
- Tang, M.; Baez, S.; Pruyas, M.; Diaz, A.; Calvo, A.; Riquelme, E.; Wistuba, I.I. Mitochondrial DNA mutation at the D310 (displacement loop) mononucleotide sequence in the pathogenesis of gallbladder carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 1041–1046. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Platek, M.; Mahasneh, A.; Ambrosone, C.B.; Zhao, H. Mitochondrial copy number and risk of breast cancer: A pilot study. Mitochondrion 2010, 10, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Alhomidi, M.A.; Vedicherla, B.; Movva, S.; Rao, P.K.; Ahuja, Y.R.; Hasan, Q. Mitochondrial D310 instability in Asian Indian breast cancer patients. Tumour Biol. J. Int. Soc. Oncodevelopmental. Biol. Med. 2013, 34, 2427–2432. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, H.; Hattori, K.; Wada, R.; Ishikawa, K.; Fukuda, S.; Takenaga, K.; Nakada, K.; Hayashi, J. Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PLoS ONE 2011, 6, e23401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, R.-K.; Chang, J.; Yeh, K.-T.; Lou, M.A.; Lu, J.-F.; Tan, D.-J.; Liu, H.; Wong, L.-J.C. Mitochondrial DNA content varies with pathological characteristics of breast cancer. J. Oncol. 2011, 2011, 496189. [Google Scholar] [CrossRef] [Green Version]
- Czarnecka, A.M.; Krawczyk, T.; Zdrozny, M.; Lubiński, J.; Arnold, R.S.; Kukwa, W.; Scińska, A.; Golik, P.; Bartnik, E.; Petros, J.A. Mitochondrial NADH-dehydrogenase subunit 3 (ND3) polymorphism (A10398G) and sporadic breast cancer in Poland. Breast Cancer Res. Treat. 2010, 121, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, A.M.; Krawczyk, T.; Plak, K.; Klemba, A.; Zdrozny, M.; Arnold, R.S.; Kofler, B.; Golik, P.; Szybinska, A.; Lubinski, J.; et al. Mitochondrial genotype and breast cancer predisposition. Oncol. Rep. 2010, 24, 1521–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vitto, H.; Mendonça, B.S.; Elseth, K.M.; Vesper, B.J.; Portari, E.A.; Gallo, C.V.M.; Paradise, W.A.; Rumjanek, F.D.; Radosevich, J.A. Part II. Mitochondrial mutational status of high nitric oxide adapted cell line BT-20 (BT-20-HNO) as it relates to human primary breast tumors. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2013, 34, 337–347. [Google Scholar] [CrossRef]
- Fang, H.; Shen, L.; Chen, T.; He, J.; Ding, Z.; Wei, J.; Qu, J.; Chen, G.; Lu, J.; Bai, Y. Cancer type-specific modulation of mitochondrial haplogroups in breast, colorectal and thyroid cancer. BMC Cancer 2010, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez Povedano, C.; Salgado, J.; Gil, C.; Robles, M.; Patiño-García, A.; García-Foncillas, J. Analysis of BRCA1 and mtDNA haplotypes and mtDNA polymorphism in familial breast cancer. Mitochondrial DNA 2015, 26, 227–231. [Google Scholar] [CrossRef]
- Kuo, S.-J.; Chen, M.; Ma, G.-C.; Chen, S.-T.; Chang, S.-P.; Lin, W.-Y.; Chen, Y.-C.; Lee, T.-H.; Lin, T.-T.; Liu, C.-S. Number of somatic mutations in the mitochondrial D-loop region indicates poor prognosis in breast cancer, independent of TP53 mutation. Cancer Genet. Cytogenet. 2010, 201, 94–101. [Google Scholar] [CrossRef]
- Nie, H.; Shu, H.; Vartak, R.; Milstein, A.C.; Mo, Y.; Hu, X.; Fang, H.; Shen, L.; Ding, Z.; Lu, J.; et al. Mitochondrial common deletion, a potential biomarker for cancer occurrence, is selected against in cancer background: A meta-analysis of 38 studies. PLoS ONE 2013, 8, e67953. [Google Scholar] [CrossRef] [Green Version]
- Rahmani, B.; Azimi, C.; Omranipour, R.; Raoofian, R.; Zendehdel, K.; Saee-Rad, S.; Heidari, M. Mutation screening in the mitochondrial D-loop region of tumoral and non-tumoral breast cancer in Iranian patients. Acta Med. Iran. 2012, 50, 447–453. [Google Scholar] [PubMed]
- Rohan, T.E.; Wong, L.-J.; Wang, T.; Haines, J.; Kabat, G.C. Do alterations in mitochondrial DNA play a role in breast carcinogenesis? J. Oncol. 2010, 2010, 604304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultana, G.N.N.; Rahman, A.; Shahinuzzaman, A.D.A.; Begum, R.A.; Hossain, C.F. Mitochondrial DNA mutations---candidate biomarkers for breast cancer diagnosis in Bangladesh. Chin. J. Cancer 2012, 31, 449–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tengku Baharudin, N.; Jaafar, H.; Zainuddin, Z. Association of mitochondrial DNA 10398 polymorphism in invasive breast cancer in malay population of peninsular malaysia. Malays. J. Med. Sci. MJMS 2012, 19, 36–42. [Google Scholar]
- Thyagarajan, B.; Wang, R.; Nelson, H.; Barcelo, H.; Koh, W.-P.; Yuan, J.-M. Mitochondrial DNA copy number is associated with breast cancer risk. PLoS ONE 2013, 8, e65968. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Tran-Thanh, D.; Ma, C.; May, K.; Jung, J.; Vecchiarelli, J.; Done, S.J. Mitochondrial D310 mutations in the early development of breast cancer. Br. J. Cancer 2012, 106, 1506–1511. [Google Scholar] [CrossRef]
- Canter, J.A.; Kallianpur, A.R.; Parl, F.F.; Millikan, R.C. Mitochondrial DNA G10398A polymorphism and invasive breast cancer in African-American women. Cancer Res. 2005, 65, 8028–8033. [Google Scholar] [CrossRef] [Green Version]
- Darvishi, K.; Sharma, S.; Bhat, A.K.; Rai, E.; Bamezai, R.N.K. Mitochondrial DNA G10398A polymorphism imparts maternal Haplogroup N a risk for breast and esophageal cancer. Cancer Lett. 2007, 249, 249–255. [Google Scholar] [CrossRef]
- Zhu, W.; Qin, W.; Bradley, P.; Wessel, A.; Puckett, C.L.; Sauter, E.R. Mitochondrial DNA mutations in breast cancer tissue and in matched nipple aspirate fluid. Carcinogenesis 2005, 26, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Tseng, L.-M.; Yin, P.-H.; Chi, C.-W.; Hsu, C.-Y.; Wu, C.-W.; Lee, L.-M.; Wei, Y.-H.; Lee, H.-C. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes. Chromosomes Cancer 2006, 45, 629–638. [Google Scholar] [CrossRef]
- Brandon, M.; Baldi, P.; Wallace, D.C. Mitochondrial mutations in cancer. Oncogene 2006, 25, 4647–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzybowska-Szatkowska, L.; Slaska, B. Polymorphisms in genes encoding mt-tRNA in female breast cancer in Poland. Mitochondrial DNA 2012, 23, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Santidrian, A.F.; Matsuno-Yagi, A.; Ritland, M.; Seo, B.B.; LeBoeuf, S.E.; Gay, L.J.; Yagi, T.; Felding-Habermann, B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J. Clin. Invest. 2013, 123, 1068–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyak, K.; Li, Y.; Zhu, H.; Lengauer, C.; Willson, J.K.; Markowitz, S.D.; Trush, M.A.; Kinzler, K.W.; Vogelstein, B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat. Genet. 1998, 20, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Fliss, M.S.; Usadel, H.; Caballero, O.L.; Wu, L.; Buta, M.R.; Eleff, S.M.; Jen, J.; Sidransky, D. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science 2000, 287, 2017–2019. [Google Scholar] [CrossRef] [Green Version]
- Cerutti, P.A. Prooxidant states and tumor promotion. Science 1985, 227, 375–381. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Sharma, L.K.; Bai, Y. Implications of mitochondrial DNA mutations and mitochondrial dysfunction in tumorigenesis. Cell Res. 2009, 19, 802–815. [Google Scholar] [CrossRef]
- Chatterjee, A.; Dasgupta, S.; Sidransky, D. Mitochondrial subversion in cancer. Cancer Prev. Res. Phila. Pa 2011, 4, 638–654. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, V.W.S.; Tsang, P.C.K.; Chiu, P.M.; Cheung, A.N.Y.; Khoo, U.S.; Nagley, P.; Ngan, H.Y.S. Microsatellite instability in mitochondrial genome of common female cancers. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2006, 16 (Suppl. 1), 259–266. [Google Scholar] [CrossRef]
- Bai, R.-K.; Leal, S.M.; Covarrubias, D.; Liu, A.; Wong, L.-J.C. Mitochondrial genetic background modifies breast cancer risk. Cancer Res. 2007, 67, 4687–4694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covarrubias, D.; Bai, R.-K.; Wong, L.-J.C.; Leal, S.M. Mitochondrial DNA variant interactions modify breast cancer risk. J. Hum. Genet. 2008, 53, 924–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzotti, A.; Kraft, P.; Hankinson, S.E.; Hunter, D.J.; Buring, J.; Cox, D.G. The mitochondrial A10398G polymorphism, interaction with alcohol consumption, and breast cancer risk. PLoS ONE 2009, 4, e5356. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.; Koul, A.; Sharma, S.; Rai, E.; Bukhari, S.I.A.; Dhar, M.K.; Bamezai, R.N.K. The possible role of 10398A and 16189C mtDNA variants in providing susceptibility to T2DM in two North Indian populations: A replicative study. Hum. Genet. 2007, 120, 821–826. [Google Scholar] [CrossRef]
- Tipirisetti, N.R.; Lakshmi, R.K.; Govatati, S.; Govatati, S.; Vuree, S.; Singh, L.; Raghunadha Rao, D.; Bhanoori, M.; Vishnupriya, S. Mitochondrial genome variations in advanced stage breast cancer: A case-control study. Mitochondrion 2013, 13, 372–378. [Google Scholar] [CrossRef]
- Gochhait, S.; Bhatt, A.; Sharma, S.; Singh, Y.P.; Gupta, P.; Bamezai, R.N.K. Concomitant presence of mutations in mitochondrial genome and p53 in cancer development—A study in north Indian sporadic breast and esophageal cancer patients. Int. J. Cancer 2008, 123, 2580–2586. [Google Scholar] [CrossRef]
- Tan, D.-J.; Bai, R.-K.; Wong, L.-J.C. Comprehensive scanning of somatic mitochondrial DNA mutations in breast cancer. Cancer Res. 2002, 62, 972–976. [Google Scholar]
- Ye, C.; Shu, X.O.; Pierce, L.; Wen, W.; Courtney, R.; Gao, Y.-T.; Zheng, W.; Cai, Q. Mutations in the mitochondrial DNA D-loop region and breast cancer risk. Breast Cancer Res. Treat. 2010, 119, 431–436. [Google Scholar] [CrossRef]
- Cai, F.F.; Kohler, C.; Zhang, B.; Chen, W.J.; Barekati, Z.; Garritsen, H.S.P.; Lenner, P.; Toniolo, P.; Zhang, J.J.; Zhong, X.Y. Mutations of mitochondrial DNA as potential biomarkers in breast cancer. Anticancer Res. 2011, 31, 4267–4271. [Google Scholar]
- Tseng, L.-M.; Yin, P.-H.; Yang, C.-W.; Tsai, Y.-F.; Hsu, C.-Y.; Chi, C.-W.; Lee, H.-C. Somatic mutations of the mitochondrial genome in human breast cancers. Genes. Chromosomes Cancer 2011, 50, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Fendt, L.; Niederstätter, H.; Huber, G.; Zelger, B.; Dünser, M.; Seifarth, C.; Röck, A.; Schäfer, G.; Klocker, H.; Parson, W. Accumulation of mutations over the entire mitochondrial genome of breast cancer cells obtained by tissue microdissection. Breast Cancer Res. Treat. 2011, 128, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Barekati, Z.; Radpour, R.; Kohler, C.; Zhang, B.; Toniolo, P.; Lenner, P.; Lv, Q.; Zheng, H.; Zhong, X.Y. Methylation profile of TP53 regulatory pathway and mtDNA alterations in breast cancer patients lacking TP53 mutations. Hum. Mol. Genet. 2010, 19, 2936–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platek, M.E.; Shields, P.G.; Tan, D.; Marian, C.; Bonner, M.R.; McCann, S.E.; Nie, J.; Wilding, G.E.; Ambrosone, C.; Millen, A.E.; et al. Alcohol consumption and breast tumor mitochondrial DNA mutations. Breast Cancer Res. Treat. 2010, 121, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.-Y.; Wang, H.-W.; Yao, Y.-G.; Kong, Q.-P.; Zhang, Y.-P. Somatic mutations of mitochondrial genome in early stage breast cancer. Int. J. Cancer 2007, 121, 1253–1256. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.-G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef] [Green Version]
- Horai, S.; Hayasaka, K. Intraspecific nucleotide sequence differences in the major noncoding region of human mitochondrial DNA. Am. J. Hum. Genet. 1990, 46, 828–842. [Google Scholar]
- Lee, H.-C.; Li, S.-H.; Lin, J.-C.; Wu, C.-C.; Yeh, D.-C.; Wei, Y.-H. Somatic mutations in the D-loop and decrease in the copy number of mitochondrial DNA in human hepatocellular carcinoma. Mutat. Res. 2004, 547, 71–78. [Google Scholar] [CrossRef]
- Bianchi, M.; Bianchi, N.; Bailliet, G. Mitochondrial DNA mutations in normal and tumor tissues from breast cancer patients. Cytogenet. Genome Res. 1995, 71, 99–103. [Google Scholar] [CrossRef]
- Ye, C.; Shu, X.-O.; Wen, W.; Pierce, L.; Courtney, R.; Gao, Y.-T.; Zheng, W.; Cai, Q. Quantitative analysis of mitochondrial DNA 4977-bp deletion in sporadic breast cancer and benign breast diseases. Breast Cancer Res. Treat. 2007, 108, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Hossein, R.; Houshmand, M. Diagnostic algorithm for identification of individuals with hereditary predisposition to breast cancer. Lik. Sprava. 2008, 2, 103–108. [Google Scholar]
- Weigl, S.; Paradiso, A.; Tommasi, S. Mitochondria and Familial Predisposition to Breast Cancer. Curr. Genom. 2013, 14, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Huang, P. Mitochondrial defects in cancer. Mol. Cancer 2002, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-C.; Wei, Y.-H. Mitochondrial DNA instability and metabolic shift in human cancers. Int. J. Mol. Sci. 2009, 10, 674–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.-W.; Yin, P.-H.; Lee, H.-C.; Chi, C.-W.; Tseng, L.-M. Mitochondrial DNA content as a potential marker to predict response to anthracycline in breast cancer patients. Breast J. 2010, 16, 264–270. [Google Scholar] [CrossRef]
- Chandra, D.; Singh, K.K. Genetic insights into OXPHOS defect and its role in cancer. Biochim. Biophys. Acta 2011, 1807, 620–625. [Google Scholar] [CrossRef] [Green Version]
- Kulawiec, M.; Ayyasamy, V.; Singh, K.K. p53 regulates mtDNA copy number and mitocheckpoint pathway. J. Carcinog. 2009, 8, 8. [Google Scholar] [CrossRef]
- Singh, K.K. Mitochondria damage checkpoint, aging, and cancer. Ann. N. Y. Acad. Sci. 2006, 1067, 182–190. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer: Warburg addressed. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Kaipparettu, B.A.; Ma, Y.; Park, J.H.; Lee, T.-L.; Zhang, Y.; Yotnda, P.; Creighton, C.J.; Chan, W.-Y.; Wong, L.-J.C. Crosstalk from non-cancerous mitochondria can inhibit tumor properties of metastatic cells by suppressing oncogenic pathways. PLoS ONE 2013, 8, e61747. [Google Scholar] [CrossRef] [Green Version]
- Desouki, M.M.; Kulawiec, M.; Bansal, S.; Das, G.M.; Singh, K.K. Cross talk between mitochondria and superoxide generating NADPH oxidase in breast and ovarian tumors. Cancer Biol. Ther. 2005, 4, 1367–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.K.; Kulawiec, M.; Still, I.; Desouki, M.M.; Geradts, J.; Matsui, S.-I. Inter-genomic cross talk between mitochondria and the nucleus plays an important role in tumorigenesis. Gene 2005, 354, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Bai, R.-K.; Trieu, R.; Wong, L.-J.C. Mitochondrial dysfunction in human breast cancer cells and their transmitochondrial cybrids. Biochim. Biophys. Acta 2010, 1797, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.H.; Zhou, M.; Liu, H.; Ding, Y.; Khong, H.T.; Yu, D.; Fodstad, O.; Tan, M. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 2009, 28, 3689–3701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsanou, E.; Ioachim, E.; Briasoulis, E.; Damala, K.; Charchanti, A.; Karavasilis, V.; Pavlidis, N.; Agnantis, N.J. Immunohistochemical expression of superoxide dismutase (MnSOD) anti-oxidant enzyme in invasive breast carcinoma. Histol. Histopathol. 2004, 19, 807–813. [Google Scholar] [PubMed]
- Tsai, S.-M.; Hou, M.-F.; Wu, S.-H.; Hu, B.-W.; Yang, S.-F.; Chen, W.-T.; Chai, C.-Y.; Ma, H.; Tsai, L.-Y. Expression of manganese superoxide dismutase in patients with breast cancer. Kaohsiung J. Med. Sci. 2011, 27, 167–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Voloboueva, L.A.; Ouyang, Y.; Emery, J.F.; Giffard, R.G. Overexpression of mitochondrial Hsp70/Hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J. Cereb. Blood. Flow. Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2009, 29, 365–374. [Google Scholar] [CrossRef]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 2007, 131, 257–270. [Google Scholar] [CrossRef]
- Ghosh, J.C.; Siegelin, M.D.; Dohi, T.; Altieri, D.C. Heat Shock Protein 60 Regulation of the Mitochondrial Permeability Transition Pore in Tumor Cells. Cancer Res. 2010, 70, 8988–8993. [Google Scholar] [CrossRef] [Green Version]
- Xiang, F.; Huang, Y.S.; Shi, X.H.; Zhang, Q. Mitochondrial chaperone tumour necrosis factor receptor-associated protein 1 protects cardiomyocytes from hypoxic injury by regulating mitochondrial permeability transition pore opening. FEBS J. 2010, 277, 1929–1938. [Google Scholar] [CrossRef]
- Rui, Z.; Jian-Guo, J.; Yuan-Peng, T.; Hai, P.; Bing-Gen, R. Use of serological proteomic methods to find biomarkers associated with breast cancer. Proteomics 2003, 3, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Straume, O.; Shimamura, T.; Lampa, M.J.G.; Carretero, J.; Øyan, A.M.; Jia, D.; Borgman, C.L.; Soucheray, M.; Downing, S.R.; Short, S.M.; et al. Suppression of heat shock protein 27 induces long-term dormancy in human breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 8699–8704. [Google Scholar] [CrossRef] [Green Version]
- Pfanner, N.; Wiedemann, N. Mitochondrial protein import: Two membranes, three translocases. Curr. Opin. Cell Biol. 2002, 14, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Philp, N.J.; Pestell, R.G.; Lisanti, M.P. Mitochondrial metabolism in cancer metastasis: Visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle Georget. Tex 2012, 11, 1445–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Qiao, M.; Zhang, Y.; Jiang, Y.; Wei, P.; Yao, J.; Gu, B.; Wang, Y.; Lu, J.; Wang, Z.; et al. Quantitative proteomics study of breast cancer cell lines isolated from a single patient: Discovery of TIMM17A as a marker for breast cancer. Proteomics 2010, 10, 1374–1390. [Google Scholar] [CrossRef] [PubMed]
- Salhab, M.; Patani, N.; Jiang, W.; Mokbel, K. High TIMM17A expression is associated with adverse pathological and clinical outcomes in human breast cancer. Breast Cancer Tokyo Jpn. 2012, 19, 153–160. [Google Scholar] [CrossRef]
- Han, Z.; Slack, R.S.; Li, W.; Papadopoulos, V. Expression of peripheral benzodiazepine receptor (PBR) in human tumors: Relationship to breast, colorectal, and prostate tumor progression. J. Recept. Signal Transduct. Res. 2003, 23, 225–238. [Google Scholar] [CrossRef]
- Gasparre, G.; Porcelli, A.M.; Bonora, E.; Pennisi, L.F.; Toller, M.; Iommarini, L.; Ghelli, A.; Moretti, M.; Betts, C.M.; Martinelli, G.N.; et al. Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 9001–9006. [Google Scholar] [CrossRef] [Green Version]
- Putignani, L.; Raffa, S.; Pescosolido, R.; Rizza, T.; Del Chierico, F.; Leone, L.; Aimati, L.; Signore, F.; Carrozzo, R.; Callea, F.; et al. Preliminary evidences on mitochondrial injury and impaired oxidative metabolism in breast cancer. Mitochondrion 2012, 12, 363–369. [Google Scholar] [CrossRef]
- Avagliano, A.; Ruocco, M.R.; Aliotta, F.; Belviso, I.; Accurso, A.; Masone, S.; Montagnani, S.; Arcucci, A. Mitochondrial Flexibility of Breast Cancers: A Growth Advantage and a Therapeutic Opportunity. Cells 2019, 8, 401. [Google Scholar] [CrossRef] [Green Version]
- Zou, P.; Liu, L.; Zheng, L.D.; Payne, K.K.; Manjili, M.H.; Idowu, M.O.; Zhang, J.; Schmelz, E.M.; Cheng, Z. Coordinated Upregulation of Mitochondrial Biogenesis and Autophagy in Breast Cancer Cells: The Role of Dynamin Related Protein-1 and Implication for Breast Cancer Treatment. Oxid. Med. Cell. Longev. 2016, 2016, 4085727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjaerulff, O.; Brodin, L.; Jung, A. The structure and function of endophilin proteins. Cell Biochem. Biophys. 2011, 60, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Kannan, A.; Wells, R.B.; Sivakumar, S.; Komatsu, S.; Singh, K.P.; Samten, B.; Philley, J.V.; Sauter, E.R.; Ikebe, M.; Idell, S.; et al. Mitochondrial Reprogramming Regulates Breast Cancer Progression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3348–3360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, B.K.; Deng, C.-X. Characterization of potential driver mutations involved in human breast cancer by computational approaches. Oncotarget 2017, 8, 50252–50272. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Qiao, W.; Linke, S.P.; Cao, L.; Li, W.M.; Furth, P.A.; Harris, C.C.; Deng, C.X. Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat. Genet. 2001, 28, 266–271. [Google Scholar] [CrossRef]
- Osborne, C.; Wilson, P.; Tripathy, D. Oncogenes and tumor suppressor genes in breast cancer: Potential diagnostic and therapeutic applications. Oncologist 2004, 9, 361–377. [Google Scholar] [CrossRef]
- Burke, W.; Petersen, G.; Lynch, P.; Botkin, J.; Daly, M.; Garber, J.; Kahn, M.J.; McTiernan, A.; Offit, K.; Thomson, E.; et al. Recommendations for follow-up care of individuals with an inherited predisposition to cancer. I. Hereditary nonpolyposis colon cancer. Cancer Genetics Studies Consortium. JAMA 1997, 277, 915–919. [Google Scholar] [CrossRef]
- Kerangueven, F.; Essioux, L.; Dib, A.; Noguchi, T.; Allione, F.; Geneix, J.; Longy, M.; Lidereau, R.; Eisinger, F.; Pébusque, M.J. Loss of heterozygosity and linkage analysis in breast carcinoma: Indication for a putative third susceptibility gene on the short arm of chromosome 8. Oncogene 1995, 10, 1023–1026. [Google Scholar]
- Poumpouridou, N.; Kroupis, C. Hereditary breast cancer: Beyond BRCA genetic analysis; PALB2 emerges. Clin. Chem. Lab. Med. 2011, 50, 423–434. [Google Scholar] [CrossRef]
- Ford, D.; Easton, D.F.; Stratton, M.; Narod, S.; Goldgar, D.; Devilee, P.; Bishop, D.T.; Weber, B.; Lenoir, G.; Chang-Claude, J.; et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am. J. Hum. Genet. 1998, 62, 676–689. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Beeghly-Fadiel, A.; Long, J.; Zheng, W. Genetic variants associated with breast-cancer risk: Comprehensive research synopsis, meta-analysis, and epidemiological evidence. Lancet Oncol. 2011, 12, 477–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cipollini, G.; Tommasi, S.; Paradiso, A.; Aretini, P.; Bonatti, F.; Brunetti, I.; Bruno, M.; Lombardi, G.; Schittulli, F.; Sensi, E.; et al. Genetic alterations in hereditary breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2004, 15 (Suppl. 1), I7–I13. [Google Scholar] [CrossRef] [PubMed]
- Shuen, A.Y.; Foulkes, W.D. Inherited mutations in breast cancer genes--risk and response. J. Mammary Gland Biol. Neoplasia 2011, 16, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Desrichard, A.; Bidet, Y.; Uhrhammer, N.; Bignon, Y.-J. CHEK2 contribution to hereditary breast cancer in non-BRCA families. Breast Cancer Res. BCR 2011, 13, R119. [Google Scholar] [CrossRef] [Green Version]
- Easton, D.; Ford, D.; Peto, J. Inherited susceptibility to breast cancer. Cancer Surv. 1993, 18, 95–113. [Google Scholar] [PubMed]
- van der Groep, P.; van der Wall, E.; van Diest, P.J. Pathology of hereditary breast cancer. Cell. Oncol. Dordr. 2011, 34, 71–88. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Cai, H.; Wu, T.; Sobhian, B.; Huo, Y.; Alcivar, A.; Mehta, M.; Cheung, K.L.; Ganesan, S.; Kong, A.-N.T.; et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol. Cell. Biol. 2012, 32, 1506–1517. [Google Scholar] [CrossRef] [Green Version]
- Kılıç, Y.; Çelebiler, A.Ç.; Sakızlı, M. Selecting housekeeping genes as references for the normalization of quantitative PCR data in breast cancer. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2014, 16, 184–190. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, C.; Rahman, N. Genetic predisposition to breast cancer: Past, present, and future. Annu. Rev. Genom. Hum. Genet. 2008, 9, 321–345. [Google Scholar] [CrossRef]
- Takaku, M.; Grimm, S.A.; Wade, P.A. GATA3 in Breast Cancer: Tumor Suppressor or Oncogene? Gene Expr. 2015, 16, 163–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjöblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.; et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, 869–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Banerji, S.; Cibulskis, K.; Rangel-Escareno, C.; Brown, K.K.; Carter, S.L.; Frederick, A.M.; Lawrence, M.S.; Sivachenko, A.Y.; Sougnez, C.; Zou, L.; et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012, 486, 405–409. [Google Scholar] [CrossRef] [Green Version]
- Branković-Magić, M.; Dobricić, J.; Krivokuća, A. Genetics of breast cancer: Contribution of BRCA1/2 genes alterations to hereditary predisposition. Vojnosanit. Pregl. 2012, 69, 700–706. [Google Scholar] [CrossRef]
- Lee, E.Y. Tumor suppressor genes and their alterations in breast cancer. Semin. Cancer Biol. 1995, 6, 119–125. [Google Scholar] [CrossRef]
- Deng, C.-X.; Wang, R.-H. Roles of BRCA1 in DNA damage repair: A link between development and cancer. Hum. Mol. Genet. 2003, 12, R113–R123. [Google Scholar] [CrossRef] [Green Version]
- Somasundaram, K. Breast cancer gene 1 (BRCA1): Role in cell cycle regulation and DNA repair--perhaps through transcription. J. Cell. Biochem. 2003, 88, 1084–1091. [Google Scholar] [CrossRef]
- Wu, J.; Lu, L.-Y.; Yu, X. The role of BRCA1 in DNA damage response. Protein Cell 2010, 1, 117–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medema, R.H.; Macůrek, L. Checkpoint control and cancer. Oncogene 2012, 31, 2601–2613. [Google Scholar] [CrossRef] [Green Version]
- Kwong, A.; Ng, E.K.O.; Wong, C.L.P.; Law, F.B.F.; Au, T.; Wong, H.N.; Kurian, A.W.; West, D.W.; Ford, J.M.; Ma, E.S.K. Identification of BRCA1/2 founder mutations in Southern Chinese breast cancer patients using gene sequencing and high resolution DNA melting analysis. PLoS ONE 2012, 7, e43994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilato, B.; De Summa, S.; Danza, K.; Papadimitriou, S.; Zaccagna, P.; Paradiso, A.; Tommasi, S. DHPLC/SURVEYOR nuclease: A sensitive, rapid and affordable method to analyze BRCA1 and BRCA2 mutations in breast cancer families. Mol. Biotechnol. 2012, 52, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Neupert, W.; Herrmann, J.M. Translocation of Proteins into Mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef] [Green Version]
- Coene, E.D.; Hollinshead, M.S.; Waeytens, A.A.T.; Schelfhout, V.R.J.; Eechaute, W.P.; Shaw, M.K.; Van Oostveldt, P.M.V.; Vaux, D.J. Phosphorylated BRCA1 is predominantly located in the nucleus and mitochondria. Mol. Biol. Cell 2005, 16, 997–1010. [Google Scholar] [CrossRef] [Green Version]
- Maniccia, A.W.; Lewis, C.; Begum, N.; Xu, J.; Cui, J.; Chipitsyna, G.; Aysola, K.; Reddy, V.; Bhat, G.; Fujimura, Y.; et al. Mitochondrial localization, ELK-1 transcriptional regulation and growth inhibitory functions of BRCA1, BRCA1a, and BRCA1b proteins. J. Cell. Physiol. 2009, 219, 634–641. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-L.; Ha, G.-H.; Campo, L.; Denning, M.F.; Patel, T.B.; Osipo, C.; Lin, S.-Y.; Breuer, E.-K. The role of Rak in the regulation of stability and function of BRCA1. Oncotarget 2017, 8, 86799–86815. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Amleh, A.; Sun, J.; Jin, X.; McCullough, S.D.; Baer, R.; Ren, D.; Li, R.; Hu, Y. Ubiquitination and proteasome-mediated degradation of BRCA1 and BARD1 during steroidogenesis in human ovarian granulosa cells. Mol. Endocrinol. Baltim. 2007, 21, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Li, J.; Cheng, D.; Parameswaran, B.; Zhang, S.; Jiang, Z.; Yew, P.R.; Peng, J.; Ye, Q.; Hu, Y. The F-box protein FBXO44 mediates BRCA1 ubiquitination and degradation. J. Biol. Chem. 2012, 287, 41014–41022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, A.D.; Xu, H.; Baer, R. Ubiquitination and proteasomal degradation of the BRCA1 tumor suppressor is regulated during cell cycle progression. J. Biol. Chem. 2004, 279, 33909–33918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lu, G.; Li, L.; Yi, J.; Yan, K.; Wang, Y.; Zhu, B.; Kuang, J.; Lin, M.; Zhang, S.; et al. HUWE1 interacts with BRCA1 and promotes its degradation in the ubiquitin-proteasome pathway. Biochem. Biophys. Res. Commun. 2014, 444, 549–554. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M.K. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 2014, 460, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.Y.; Yi, H.-S.; Kim, H.-W.; Shong, M. Dysregulation of mitophagy in carcinogenesis and tumor progression. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, K.; Takano, N.; Yamada, Y.; Kazama, H.; Tokuhisa, M.; Hino, H.; Fujita, K.; Barroga, E.; Hiramoto, M.; Handa, H.; et al. BRCA1 degradation in response to mitochondrial damage in breast cancer cells. Sci. Rep. 2021, 11, 8735. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [Green Version]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Chen, Q.; Lei, J.H.; Bao, J.; Wang, H.; Hao, W.; Li, L.; Peng, C.; Masuda, T.; Miao, K.; Xu, J.; et al. BRCA1 Deficiency Impairs Mitophagy and Promotes Inflammasome Activation and Mammary Tumor Metastasis. Adv. Sci. Weinh. Baden-Wurtt. Ger. 2020, 7, 1903616. [Google Scholar] [CrossRef] [Green Version]
- Privat, M.; Radosevic-Robin, N.; Aubel, C.; Cayre, A.; Penault-Llorca, F.; Marceau, G.; Sapin, V.; Bignon, Y.-J.; Morvan, D. BRCA1 induces major energetic metabolism reprogramming in breast cancer cells. PLoS ONE 2014, 9, e102438. [Google Scholar] [CrossRef] [Green Version]
- Renaudin, X.; Lee, M.; Shehata, M.; Surmann, E.-M.; Venkitaraman, A.R. BRCA2 deficiency reveals that oxidative stress impairs RNaseH1 function to cripple mitochondrial DNA maintenance. Cell Rep. 2021, 36, 109478. [Google Scholar] [CrossRef]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef]
- Tan, M.; Yao, J.; Yu, D. Overexpression of the c-erbB-2 gene enhanced intrinsic metastasis potential in human breast cancer cells without increasing their transformation abilities. Cancer Res. 1997, 57, 1199–1205. [Google Scholar] [PubMed]
- Tan, M.; Li, P.; Klos, K.S.; Lu, J.; Lan, K.-H.; Nagata, Y.; Fang, D.; Jing, T.; Yu, D. ErbB2 promotes Src synthesis and stability: Novel mechanisms of Src activation that confer breast cancer metastasis. Cancer Res. 2005, 65, 1858–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.; Lan, K.-H.; Yao, J.; Lu, C.-H.; Sun, M.; Neal, C.L.; Lu, J.; Yu, D. Selective inhibition of ErbB2-overexpressing breast cancer in vivo by a novel TAT-based ErbB2-targeting signal transducers and activators of transcription 3-blocking peptide. Cancer Res. 2006, 66, 3764–3772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.; Li, P.; Sun, M.; Yin, G.; Yu, D. Upregulation and activation of PKC alpha by ErbB2 through Src promotes breast cancer cell invasion that can be blocked by combined treatment with PKC alpha and Src inhibitors. Oncogene 2006, 25, 3286–3295. [Google Scholar] [CrossRef] [Green Version]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Zhou, X.; Tan, M.; Stone Hawthorne, V.; Klos, K.S.; Lan, K.-H.; Yang, Y.; Yang, W.; Smith, T.L.; Shi, D.; Yu, D. Activation of the Akt/mammalian target of rapamycin/4E-BP1 pathway by ErbB2 overexpression predicts tumor progression in breast cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 6779–6788. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Berezov, A.; Wang, Q.; Zhang, G.; Drebin, J.; Murali, R.; Greene, M.I. ErbB receptors: From oncogenes to targeted cancer therapies. J. Clin. Investig. 2007, 117, 2051–2058. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Liu, Z.; Desai, S.; Zhao, Y.; Liu, H.; Pannell, L.K.; Yi, H.; Wright, E.R.; Owen, L.B.; Dean-Colomb, W.; et al. Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nat. Commun. 2012, 3, 1271. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, L.; Peng, Y.; Chen, Y.; Lv, X.; Li, S.; Qin, X.; Yang, H.; Wu, C.; Liu, Y. Surface chemistry induces mitochondria-mediated apoptosis of breast cancer cells via PTEN/PI3K/AKT signaling pathway. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 172–185. [Google Scholar] [CrossRef]
- McCluggage, W.G.; Singh, N.; Gilks, C.B. Key changes to the World Health Organization (WHO) classification of female genital tumours introduced in the 5th edition (2020). Histopathology 2022, 80, 762–778. [Google Scholar] [CrossRef]
- Rodriguez, A.C.; Blanchard, Z.; Maurer, K.A.; Gertz, J. Estrogen Signaling in Endometrial Cancer: A Key Oncogenic Pathway with Several Open Questions. Horm. Cancer 2019, 10, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosbie, E.J.; Kitson, S.J.; McAlpine, J.N.; Mukhopadhyay, A.; Powell, M.E.; Singh, N. Endometrial cancer. Lancet 2022, 399, 1412–1428. [Google Scholar] [CrossRef] [PubMed]
- Mahdy, H.; Casey, M.J.; Crotzer, D. Endometrial Cancer. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Okuda, T.; Sekizawa, A.; Purwosunu, Y.; Nagatsuka, M.; Morioka, M.; Hayashi, M.; Okai, T. Genetics of Endometrial Cancers. Obstet. Gynecol. Int. 2010, 2010, e984013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ollikainen, M.; Abdel-Rahman, W.M.; Moisio, A.-L.; Lindroos, A.; Kariola, R.; Järvelä, I.; Pöyhönen, M.; Butzow, R.; Peltomäki, P. Molecular analysis of familial endometrial carcinoma: A manifestation of hereditary nonpolyposis colorectal cancer or a separate syndrome? J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 4609–4616. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knez, J.; Al Mahdawi, L.; Takač, I.; Sobočan, M. The Perspectives of Fertility Preservation in Women with Endometrial Cancer. Cancers 2021, 13, 602. [Google Scholar] [CrossRef]
- Jeske, Y.W.; Ali, S.; Byron, S.A.; Gao, F.; Mannel, R.S.; Ghebre, R.G.; DiSilvestro, P.A.; Lele, S.B.; Pearl, M.L.; Schmidt, A.P.; et al. FGFR2 mutations are associated with poor outcomes in endometrioid endometrial cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2017, 145, 366–373. [Google Scholar] [CrossRef] [Green Version]
- Kong, A.; Johnson, N.; Kitchener, H.C.; Lawrie, T.A. Adjuvant radiotherapy for stage I endometrial cancer. Cochrane Database Syst. Rev. 2012, 18, CD003916. [Google Scholar]
- Chiu, W.K.; Kwok, S.T.; Wang, Y.; Luk, H.M.; Chan, A.H.Y.; Tse, K.Y. Applications and Safety of Sentinel Lymph Node Biopsy in Endometrial Cancer. J. Clin. Med. 2022, 11, 6462. [Google Scholar] [CrossRef]
- Kok, P.S.; Antill, Y.C.; Scott, C.L.; Lee, C.K. The impact of single agent PD-1 or PD-L1 inhibition on advanced endometrial cancers: Meta-analysis. ESMO Open. 2022, 18, 100635. [Google Scholar] [CrossRef]
- Huang, P.; Fan, X.; Yu, H.; Zhang, K.; Li, H.; Wang, Y.; Xue, F. Glucose metabolic reprogramming and its therapeutic potential in obesity-associated endometrial cancer. J. Transl. Med. 2023, 21, 94. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Hatakeyama, K.; Nagashima, T.; Ohshima, K.; Urakami, K.; Yamaguchi, K.; Hirashima, Y. Activation of oxidative phosphorylation in TP53-inactive endometrial carcinomas with a poor prognosis. Int. J. Gynecol. Cancer 2021, 31, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Somuncu, B.; Ekmekcioglu, A.; Antmen, F.M.; Ertuzun, T.; Deniz, E.; Keskin, N.; Park, J.; Yazici, I.E.; Simsek, B.; Erman, B.; et al. Targeting mitochondrial DNA polymerase gamma for selective inhibition of MLH1 deficient colon cancer growth. PLoS ONE 2022, 17, e0268391. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Kurelac, I.; Cormio, A.; Zuntini, R.; Amato, L.B.; Ceccarelli, C.; Santini, D.; Cormio, G.; Fracasso, F.; Selvaggi, L.; et al. Placing mitochondrial DNA mutations within the progression model of type I endometrial carcinoma. Hum. Mol. Genet. 2011, 20, 2394–2405. [Google Scholar] [CrossRef]
- Liu, J.; Chen, T.; Yang, M.; Zhong, Z.; Ni, S.; Yang, S.; Shao, F.; Cai, L.; Bai, J.; Yu, H. Development of an Oxidative Phosphorylation-Related and Immune Microenvironment Prognostic Signature in Uterine Corpus Endometrial Carcinoma. Front. Cell Dev. Biol. 2021, 9, 753004. [Google Scholar] [CrossRef]
- Byrne, F.L.; Poon, I.K.; Modesitt, S.C.; Tomsig, J.L.; Chow, J.D.; Healy, M.E.; Baker, W.D.; Atkins, K.A.; Lancaster, J.M.; Marchion, D.C.; et al. Metabolic Vulnerabilities in Endometrial Cancer. Cancer Res. 2014, 74, 5832–5845. [Google Scholar] [CrossRef] [Green Version]
- Reznik, E.; Miller, M.L.; Şenbabaoğlu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A. Mitochondrial DNA copy number variation across human cancers. eLife 2016, 5, e10769. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, V.W.S.; Xue, W.C.; Tsang, P.C.K.; Cheung, A.N.Y.; Ngan, H.Y.S. The increase of mitochondrial DNA content in endometrial adenocarcinoma cells: A quantitative study using laser-captured microdissected tissues. Gynecol. Oncol. 2005, 98, 104–110. [Google Scholar] [CrossRef]
- Cormio, A.; Guerra, F.; Cormio, G.; Pesce, V.; Fracasso, F.; Loizzi, V.; Cantatore, P.; Selvaggi, L.; Gadaleta, M.N. The PGC-1alpha-dependent pathway of mitochondrial biogenesis is upregulated in type I endometrial cancer. Biochem. Biophys. Res. Commun. 2009, 390, 1182–1185. [Google Scholar] [CrossRef]
- Wersäll, O.C.; Löfstedt, L.; Govorov, I.; Mints, M.; Gabrielson, M.; Shoshan, M. PGC1α and VDAC1 expression in endometrial cancer. Mol. Clin. Oncol. 2021, 14, 42. [Google Scholar] [CrossRef]
- Cormio, A.; Musicco, C.; Gasparre, G.; Cormio, G.; Pesce, V.; Sardanelli, A.M.; Gadaleta, M.N. Increase in proteins involved in mitochondrial fission, mitophagy, proteolysis and antioxidant response in type I endometrial cancer as an adaptive response to respiratory complex I deficiency. Biochem. Biophys. Res. Commun. 2017, 491, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Semczuk, A.; Lorenc, A.; Putowski, L.; Futyma, K.; Bryk, J.; Miotla, P.; Bartnik, E. Clinicoprognostical features of endometrial cancer patients with somatic mtDNA mutations. Oncol. Rep. 2006, 16, 1041–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, V.W.S.; Wang, Y.; Yang, H.-J.; Tsang, P.C.K.; Ng, T.-Y.; Wong, L.-C.; Nagley, P.; Ngan, H.Y.S. Mitochondrial DNA variant 16189T>C is associated with susceptibility to endometrial cancer. Hum. Mutat. 2003, 22, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hu, Y.; Chen, B.; Tang, W.; Han, X.; Yu, H.; Xiao, C. Mitochondrial polymorphisms as risk factors for endometrial cancer in southwest China. Int. J. Gynecol. Cancer 2006, 16, 1661–1667. [Google Scholar] [CrossRef]
- Musicco, C.; Cormio, G.; Pesce, V.; Loizzi, V.; Cicinelli, E.; Resta, L.; Ranieri, G.; Cormio, A. Mitochondrial Dysfunctions in Type I Endometrial Carcinoma: Exploring Their Role in Oncogenesis and Tumor Progression. Int. J. Mol. Sci. 2018, 19, 2076. [Google Scholar] [CrossRef] [Green Version]
- Nero, C.; Ciccarone, F.; Pietragalla, A.; Scambia, G. PTEN and Gynecological Cancers. Cancers 2019, 11, 1458. [Google Scholar] [CrossRef] [Green Version]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Martins, F.C.; Couturier, D.-L.; Paterson, A.; Karnezis, A.N.; Chow, C.; Nazeran, T.M.; Odunsi, A.; Gentry-Maharaj, A.; Vrvilo, A.; Hein, A.; et al. Clinical and pathological associations of PTEN expression in ovarian cancer: A multicentre study from the Ovarian Tumour Tissue Analysis Consortium. Br. J. Cancer 2020, 123, 793–802. [Google Scholar] [CrossRef]
- Luongo, F.; Colonna, F.; Calapà, F.; Vitale, S.; Fiori, M.E.; De Maria, R. PTEN Tumor-Suppressor: The Dam of Stemness in Cancer. Cancers 2019, 11, 1076. [Google Scholar] [CrossRef] [Green Version]
- Wolff, A.C.; Domchek, S.M.; Davidson, N.E.; Sacchini, V.; McCormick, B. Cancer of the Breast. In Abeloff’s Clinical Oncology; Elsevier: Amsterdam, The Netherlands, 2014; pp. 1630–1692.e9. ISBN 978-1-4557-2865-7. [Google Scholar]
- Carnero, A.; Paramio, J.M. The PTEN/PI3K/AKT Pathway in vivo, Cancer Mouse Models. Front. Oncol. 2014, 4, 252. [Google Scholar] [CrossRef] [Green Version]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Hoell, P.; Ahlemeyer, B.; Krieglstein, J. PTEN: A crucial mediator of mitochondria-dependent apoptosis. Apoptosis Int. J. Program. Cell Death 2006, 11, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Li, R.; Xu, X.; Zhang, L.; Lian, R.; Fang, L.; Huang, Y.; Feng, X.; Liu, X.; Li, X.; et al. CK1α suppresses lung tumour growth by stabilizing PTEN and inducing autophagy. Nat. Cell Biol. 2018, 20, 465–478. [Google Scholar] [CrossRef]
- Ueno, T.; Sato, W.; Horie, Y.; Komatsu, M.; Tanida, I.; Yoshida, M.; Ohshima, S.; Mak, T.W.; Watanabe, S.; Kominami, E. Loss of Pten, a tumor suppressor, causes the strong inhibition of autophagy without affecting LC3 lipidation. Autophagy 2008, 4, 692–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2001, 276, 35243–35246. [Google Scholar] [CrossRef] [Green Version]
- Errafiy, R.; Aguado, C.; Ghislat, G.; Esteve, J.M.; Gil, A.; Loutfi, M.; Knecht, E. PTEN Increases Autophagy and Inhibits the Ubiquitin-Proteasome Pathway in Glioma Cells Independently of its Lipid Phosphatase Activity. PLoS ONE 2013, 8, e83318. [Google Scholar] [CrossRef]
- Li, M.; Yang, X.; Wang, S. PTEN enhances nasal epithelial cell resistance to TNFα-induced inflammatory injury by limiting mitophagy via repression of the TLR4-JNK-Bnip3 pathway. Mol. Med. Rep. 2018, 18, 2973–2986. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Lu, G.; Shen, H.-M. The Long and the Short of PTEN in the Regulation of Mitophagy. Front. Cell Dev. Biol. 2020, 8, 299. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef] [Green Version]
- Serebriiskii, I.G.; Pavlov, V.; Tricarico, R.; Andrianov, G.; Nicolas, E.; Parker, M.I.; Newberg, J.; Frampton, G.; Meyer, J.E.; Golemis, E.A. Comprehensive characterization of PTEN mutational profile in a series of 34,129 colorectal cancers. Nat. Commun. 2022, 13, 1618. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, D.; Longy, M. Mutations of the human PTEN gene. Hum. Mutat. 2000, 16, 109–122. [Google Scholar] [CrossRef] [PubMed]
- González-García, A.; Garrido, A.; Carrera, A.C. Targeting PTEN Regulation by Post Translational Modifications. Cancers 2022, 14, 5613. [Google Scholar] [CrossRef]
- Bhyan, S.B.; Wee, Y.; Liu, Y.; Cummins, S.; Zhao, M. Integrative analysis of common genes and driver mutations implicated in hormone stimulation for four cancers in women. PeerJ 2019, 7, e6872. [Google Scholar] [CrossRef] [Green Version]
- Cybulska, P.; Paula, A.D.C.; Tseng, J.; Leitao, M.M.; Bashashati, A.; Huntsman, D.G.; Nazeran, T.M.; Aghajanian, C.; Abu-Rustum, N.R.; DeLair, D.F.; et al. Molecular profiling and molecular classification of endometrioid ovarian carcinomas. Gynecol. Oncol. 2019, 154, 516–523. [Google Scholar] [CrossRef]
- Catalogue of Somatic Mutations in Cancer. Available online: https://cancer.sanger.ac.uk/cosmic (accessed on 10 October 2022).
- CBioportal for Cancer Genomics. Available online: http://www.cbioportal.org (accessed on 18 October 2022).
- Dinulescu, D.M.; Ince, T.A.; Quade, B.J.; Shafer, S.A.; Crowley, D.; Jacks, T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat. Med. 2005, 11, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Sherman-Baust, C.A.; Kuhn, E.; Valle, B.L.; Shih, I.-M.; Kurman, R.J.; Wang, T.-L.; Amano, T.; Ko, M.S.; Miyoshi, I.; Araki, Y.; et al. A genetically engineered ovarian cancer mouse model based on fallopian tube transformation mimics human high-grade serous carcinoma development: Mouse model of fallopian transformation develops ovarian carcinoma. J. Pathol. 2014, 233, 228–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, M.; Jin, V.; Bergsten, T.M.; Austin, J.R.; Lantvit, D.D.; Russo, A.; Burdette, J.E. Loss of PTEN in Fallopian Tube Epithelium Results in Multicellular Tumor Spheroid Formation and Metastasis to the Ovary. Cancers 2019, 11, 884. [Google Scholar] [CrossRef] [Green Version]
- Whitman, M.; Downes, C.P.; Keeler, M.; Keller, T.; Cantley, L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 1988, 332, 644–646. [Google Scholar] [CrossRef]
- Gasparri, M.; Bardhi, E.; Ruscito, I.; Papadia, A.; Farooqi, A.; Marchetti, C.; Bogani, G.; Ceccacci, I.; Mueller, M.; Benedetti Panici, P. PI3K/AKT/mTOR Pathway in Ovarian Cancer Treatment: Are We on the Right Track? Geburtshilfe Frauenheilkd. 2017, 77, 1095–1103. [Google Scholar] [CrossRef] [Green Version]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Dong, X.; Xie, S.; Zhang, L.; Zeng, P.; Zhang, L. Cellular Mechanism of Gene Mutations and Potential Therapeutic Targets in Ovarian Cancer. Cancer Manag. Res. 2021, 13, 3081–3100. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [CrossRef] [Green Version]
- Mjos, S.; Werner, H.M.J.; Birkeland, E.; Holst, F.; Berg, A.; Halle, M.K.; Tangen, I.L.; Kusonmano, K.; Mauland, K.K.; Oyan, A.M.; et al. PIK3CA exon9 mutations associate with reduced survival, and are highly concordant between matching primary tumors and metastases in endometrial cancer. Sci. Rep. 2017, 7, 10240. [Google Scholar] [CrossRef] [Green Version]
- Tondo-Steele, K.; McLean, K. The “Sweet Spot” of Targeting Tumor Metabolism in Ovarian Cancers. Cancers 2022, 14, 4696. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.D.; Han, C.C.; Wan, H.F.; He, F.; Xu, H.Y.; Wei, S.H.; Du, X.H.; Xu, F. Effects of inhibiting PI3K-Akt-mTOR pathway on lipid metabolism homeostasis in goose primary hepatocytes. Anim. Int. J. Anim. Biosci. 2016, 10, 1319–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efsun Antmen, S.; Canacankatan, N.; Gürses, İ.; Aytan, H.; Erden Ertürk, S. Relevance of lipogenesis and AMPK/Akt/mTOR signaling pathway in endometrial cancer. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Kma, L.; Baruah, T.J. The interplay of ROS and the PI3K/Akt pathway in autophagy regulation. Biotechnol. Appl. Biochem. 2022, 69, 248–264. [Google Scholar] [CrossRef]
- Soutar, M.P.M.; Kempthorne, L.; Miyakawa, S.; Annuario, E.; Melandri, D.; Harley, J.; O’Sullivan, G.A.; Wray, S.; Hancock, D.C.; Cookson, M.R.; et al. AKT signalling selectively regulates PINK1 mitophagy in SHSY5Y cells and human iPSC-derived neurons. Sci. Rep. 2018, 8, 8855. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yang, N.; Yang, C.; Ding, H.; Luo, C.; Zhang, Y.; Wu, M.; Zhang, X.; Shen, X.; Jiang, H.; et al. S9, a Novel Anticancer Agent, Exerts Its Anti-Proliferative Activity by Interfering with Both PI3K-Akt-mTOR Signaling and Microtubule Cytoskeleton. PLoS ONE 2009, 4, e4881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caino, M.C.; Ghosh, J.C.; Chae, Y.C.; Vaira, V.; Rivadeneira, D.B.; Faversani, A.; Rampini, P.; Kossenkov, A.V.; Aird, K.M.; Zhang, R.; et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc. Natl. Acad. Sci. USA 2015, 112, 8638–8643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catasus, L.; Gallardo, A.; Cuatrecasas, M.; Prat, J. PIK3CA mutations in the kinase domain (exon 20) of uterine endometrial adenocarcinomas are associated with adverse prognostic parameters. Mod. Pathol. Off. J. US Can. Acad. Pathol. Inc. 2008, 21, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasti, A.R.; Guimaraes-Young, A.; Datko, F.; Borges, V.F.; Aisner, D.L.; Shagisultanova, E. PIK3CA Mutations Drive Therapeutic Resistance in Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer. JCO Precis. Oncol. 2022, 6, e2100370. [Google Scholar] [CrossRef] [PubMed]
- Jančík, S.; Drábek, J.; Radzioch, D.; Hajdúch, M. Clinical Relevance of KRAS in Human Cancers. J. Biomed. Biotechnol. 2010, 2010, 960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias Carvalho, P.; Guimarães, C.F.; Cardoso, A.P.; Mendonça, S.; Costa, Â.M.; Oliveira, M.J.; Velho, S. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2018, 78, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Nagdas, S.; Kashatus, J.A.; Nascimento, A.; Hussain, S.S.; Trainor, R.E.; Pollock, S.R.; Adair, S.J.; Michaels, A.D.; Sesaki, H.; Stelow, E.B.; et al. Drp1 Promotes KRas-Driven Metabolic Changes to Drive Pancreatic Tumor Growth. Cell Rep. 2019, 28, 1845–1859.e5. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Nguyen, N.D.; Huang, Y.; Lin, D.; Fujimoto, T.N.; Molkentine, J.M.; Deorukhkar, A.; Kang, Y.; San Lucas, F.A.; Fernandes, C.J.; et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight 2019, 5, e126915. [Google Scholar] [CrossRef]
- Courtois, S.; de Luxán-Delgado, B.; Penin-Peyta, L.; Royo-García, A.; Parejo-Alonso, B.; Jagust, P.; Alcalá, S.; Rubiolo, J.A.; Sánchez, L.; Sainz, B.; et al. Inhibition of Mitochondrial Dynamics Preferentially Targets Pancreatic Cancer Cells with Enhanced Tumorigenic and Invasive Potential. Cancers 2021, 13, 698. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.S.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Alam, A.; Pant, R.; Chattopadhyay, S. Wnt Signaling and Its Significance Within the Tumor Microenvironment: Novel Therapeutic Insights. Front. Immunol. 2019, 10, 2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, D.L.; Weis, W.I. Beta-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat. Struct. Mol. Biol. 2005, 12, 364–371. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell. 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Ramakrishnan, A.-B.; Cadigan, K.M. Wnt target genes and where to find them. F1000Research 2017, 6, 746. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, M.L.; Ortolano, N.A.; Romero-Morales, A.I.; Gama, V. Wnt Signaling and Its Impact on Mitochondrial and Cell Cycle Dynamics in Pluripotent Stem Cells. Genes 2018, 9, 109. [Google Scholar] [CrossRef] [Green Version]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Chen, G.T.; Puttock, E.; Wang, K.; Edwards, R.A.; Waterman, M.L.; Lowengrub, J. Mathematical modeling links Wnt signaling to emergent patterns of metabolism in colon cancer. Mol. Syst. Biol. 2017, 13, 912. [Google Scholar] [CrossRef]
- Thompson, C.B. Wnt meets Warburg: Another piece in the puzzle? EMBO J. 2014, 33, 1420–1422. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Aerobic Glycolysis Hypothesis Through WNT/Beta-Catenin Pathway in Exudative Age-Related Macular Degeneration. J. Mol. Neurosci. MN 2017, 62, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Sprowl-Tanio, S.; Habowski, A.N.; Pate, K.T.; McQuade, M.M.; Wang, K.; Edwards, R.A.; Grun, F.; Lyou, Y.; Waterman, M.L. Lactate/pyruvate transporter MCT-1 is a direct Wnt target that confers sensitivity to 3-bromopyruvate in colon cancer. Cancer Metab. 2016, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Stringari, C.; Edwards, R.A.; Pate, K.T.; Waterman, M.L.; Donovan, P.J.; Gratton, E. Metabolic trajectory of cellular differentiation in small intestine by Phasor Fluorescence Lifetime Microscopy of NADH. Sci. Rep. 2012, 2, 568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senni, N.; Savall, M.; Cabrerizo Granados, D.; Alves-Guerra, M.-C.; Sartor, C.; Lagoutte, I.; Gougelet, A.; Terris, B.; Gilgenkrantz, H.; Perret, C.; et al. β-catenin-activated hepatocellular carcinomas are addicted to fatty acids. Gut 2019, 68, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Ferré, P.; Foufelle, F. SREBP-1c Transcription Factor and Lipid Homeostasis: Clinical Perspective. Horm. Res. Paediatr. 2007, 68, 72–82. [Google Scholar] [CrossRef]
- Zhang, L.; Li, S.; Wang, R.; Chen, C.; Ma, W.; Cai, H. Anti-tumor effect of LATS2 on liver cancer death: Role of DRP1-mediated mitochondrial division and the Wnt/β-catenin pathway. Biomed. Pharmacother. Biomedecine Pharmacother. 2019, 114, 108825. [Google Scholar] [CrossRef]
- Trejo-Solis, C.; Escamilla-Ramirez, A.; Jimenez-Farfan, D.; Castillo-Rodriguez, R.A.; Flores-Najera, A.; Cruz-Salgado, A. Crosstalk of the Wnt/β-Catenin Signaling Pathway in the Induction of Apoptosis on Cancer Cells. Pharmaceuticals 2021, 14, 871. [Google Scholar] [CrossRef]
- Giles, R.H.; van Es, J.H.; Clevers, H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta 2003, 1653, 1–24. [Google Scholar] [CrossRef]
- Zyla, R.E.; Olkhov-Mitsel, E.; Amemiya, Y.; Bassiouny, D.; Seth, A.; Djordjevic, B.; Nofech-Mozes, S.; Parra-Herran, C. CTNNB1 Mutations and Aberrant β-Catenin Expression in Ovarian Endometrioid Carcinoma: Correlation With Patient Outcome. Am. J. Surg. Pathol. 2021, 45, 68–76. [Google Scholar] [CrossRef]
- Kim, S.; Jeong, S. Mutation Hotspots in the β-Catenin Gene: Lessons from the Human Cancer Genome Databases. Mol. Cells 2019, 42, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Fatima, I.; Barman, S.; Rai, R.; Thiel, K.W.; Chandra, V. Targeting Wnt Signaling in Endometrial Cancer. Cancers 2021, 13, 2351. [Google Scholar] [CrossRef] [PubMed]
- Phan, P.; Saikia, B.B.; Sonnaila, S.; Agrawal, S.; Alraawi, Z.; Kumar, T.K.S.; Iyer, S. The Saga of Endocrine FGFs. Cells 2021, 10, 2418. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5, 1–38. [Google Scholar] [CrossRef]
- Chioni, A.-M.; Grose, R.P. Biological Significance and Targeting of the FGFR Axis in Cancer. Cancers 2021, 13, 5681. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/search/fgf (accessed on 8 December 2022).
- Li, X. The FGF metabolic axis. Front. Med. 2019, 13, 511–530. [Google Scholar] [CrossRef] [Green Version]
- Ligumsky, H.; Merenbakh-Lamin, K.; Keren-Khadmy, N.; Wolf, I.; Rubinek, T. The role of α-klotho in human cancer: Molecular and clinical aspects. Oncogene 2022, 41, 4487–4497. [Google Scholar] [CrossRef]
- Lojkin, I.; Rubinek, T.; Orsulic, S.; Schwarzmann, O.; Karlan, B.Y.; Bose, S.; Wolf, I. Reduced expression and growth inhibitory activity of the aging suppressor klotho in epithelial ovarian cancer. Cancer Lett. 2015, 362, 149–157. [Google Scholar] [CrossRef]
- Lu, H.; Shi, X.; Wu, G.; Zhu, J.; Song, C.; Zhang, Q.; Yang, G. FGF13 regulates proliferation and differentiation of skeletal muscle by down-regulating Spry1. Cell Prolif. 2015, 48, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Sochacka, M.; Karelus, R.; Opalinski, L.; Krowarsch, D.; Biadun, M.; Otlewski, J.; Zakrzewska, M. FGF12 is a novel component of the nucleolar NOLC1/TCOF1 ribosome biogenesis complex. Cell Commun. Signal. 2022, 20, 182. [Google Scholar] [CrossRef]
- Lee, K.W.; An, Y.J.; Lee, J.; Jung, Y.-E.; Ko, I.Y.; Jin, J.; Park, J.H.; Lee, W.K.; Cha, K.; Ko, S.-S.C.; et al. Expression and purification of intracrine human FGF 11 and study of its FGFR-dependent biological activity. J. Microbiol. Seoul Korea 2022, 60, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, V.D.; Ittmann, M.; Spencer, D.M. Paths of FGFR-driven tumorigenesis. Cell Cycle Georget. Tex 2009, 8, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Santolla, M.F.; Maggiolini, M. The FGF/FGFR System in Breast Cancer: Oncogenic Features and Therapeutic Perspectives. Cancers 2020, 12, 3029. [Google Scholar] [CrossRef] [PubMed]
- Barillari, G.; Iovane, A.; Bonuglia, M.; Albonici, L.; Garofano, P.; Di Campli, E.; Falchi, M.; Condò, I.; Manzari, V.; Ensoli, B. Fibroblast growth factor-2 transiently activates the p53 oncosuppressor protein in human primary vascular smooth muscle cells: Implications for atherogenesis. Atherosclerosis 2010, 210, 400–406. [Google Scholar] [CrossRef]
- Yang, X.; Steinberg, F.; Zhuang, L.; Bessey, R.; Trueb, B. Receptor FGFRL1 does not promote cell proliferation but induces cell adhesion. Int. J. Mol. Med. 2016, 38, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Takei, Y.; Matsumura, T.; Watanabe, K.; Nakamine, H.; Sudo, T.; Shimizu, K.; Shimada, Y. FGFRL1 deficiency reduces motility and tumorigenic potential of cells derived from oesophageal squamous cell carcinomas. Oncol. Lett. 2018, 16, 809–814. [Google Scholar] [CrossRef]
- Feng, S.; Zhou, L.; Nice, E.C.; Huang, C. Fibroblast growth factor receptors: Multifactorial-contributors to tumor initiation and progression. Histol. Histopathol. 2015, 30, 13–31. [Google Scholar] [CrossRef]
- Xie, B.; Chen, J.; Liu, B.; Zhan, J. Klotho Acts as a Tumor Suppressor in Cancers. Pathol. Oncol. Res. 2013, 19, 611–617. [Google Scholar] [CrossRef]
- Wójcik-Krowiranda, K.M.; Szczepaniec, S.; Bieńkiewicz, A. The role of the βKlotho gene in uterine endometrial cancer. Ginekol. Pol. 2018, 89, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Rubinek, T.; Shulman, M.; Israeli, S.; Bose, S.; Avraham, A.; Zundelevich, A.; Evron, E.; Gal-Yam, E.N.; Kaufman, B.; Wolf, I. Epigenetic silencing of the tumor suppressor klotho in human breast cancer. Breast Cancer Res. Treat. 2012, 133, 649–657. [Google Scholar] [CrossRef]
- Arbel Rubinstein, T.; Shahmoon, S.; Zigmond, E.; Etan, T.; Merenbakh-Lamin, K.; Pasmanik-Chor, M.; Har-Zahav, G.; Barshack, I.; Vainer, G.W.; Skalka, N.; et al. Klotho suppresses colorectal cancer through modulation of the unfolded protein response. Oncogene 2019, 38, 794–807. [Google Scholar] [CrossRef] [PubMed]
- Mäkelä, J.; Tselykh, T.V.; Maiorana, F.; Eriksson, O.; Do, H.T.; Mudò, G.; Korhonen, L.T.; Belluardo, N.; Lindholm, D. Fibroblast growth factor-21 enhances mitochondrial functions and increases the activity of PGC-1α in human dopaminergic neurons via Sirtuin-1. SpringerPlus 2014, 3, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srisakuldee, W.; Nickel, B.E.; Fandrich, R.R.; Zhang, F.; Pasumarthi, K.B.S.; Kardami, E. A Cardiac Mitochondrial FGFR1 Mediates the Antithetical Effects of FGF2 Isoforms on Permeability Transition. Cells 2021, 10, 2735. [Google Scholar] [CrossRef] [PubMed]
- Hitosugi, T.; Fan, J.; Chung, T.-W.; Lythgoe, K.; Wang, X.; Xie, J.; Ge, Q.; Gu, T.-L.; Polakiewicz, R.D.; Roesel, J.L.; et al. Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol. Cell 2011, 44, 864–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Ren, H.; Thakur, A.; Yang, T.; Li, Y.; Zhang, S.; Wang, T.; Chen, M. Decreased Level of Klotho Contributes to Drug Resistance in Lung Cancer Cells: Involving in Klotho-Mediated Cell Autophagy. DNA Cell Biol. 2016, 35, 751–757. [Google Scholar] [CrossRef]
- Zhou, H.; Pu, S.; Zhou, H.; Guo, Y. Klotho as Potential Autophagy Regulator and Therapeutic Target. Front. Pharmacol. 2021, 12, 755366. [Google Scholar] [CrossRef] [PubMed]
- Shmulevich, R.; Nissim, T.B.-K.; Wolf, I.; Merenbakh-Lamin, K.; Fishman, D.; Sekler, I.; Rubinek, T. Klotho rewires cellular metabolism of breast cancer cells through alteration of calcium shuttling and mitochondrial activity. Oncogene 2020, 39, 4636–4649. [Google Scholar] [CrossRef]
- Birrer, M.J.; Johnson, M.E.; Hao, K.; Wong, K.-K.; Park, D.-C.; Bell, A.; Welch, W.R.; Berkowitz, R.S.; Mok, S.C. Whole genome oligonucleotide-based array comparative genomic hybridization analysis identified fibroblast growth factor 1 as a prognostic marker for advanced-stage serous ovarian adenocarcinomas. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 2281–2287. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.H.; Xu, E.; Hildebrandt, M.A.T.; Liang, D.; Lu, K.; Ye, Y.; Wagar, E.A.; Wu, X. Genetic variants in the fibroblast growth factor pathway as potential markers of ovarian cancer risk, therapeutic response, and clinical outcome. Clin. Chem. 2014, 60, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Mok, S.C.; Oliva, E.; Kim, S.; Mohapatra, G.; Birrer, M.J. FGF18 as a prognostic and therapeutic biomarker in ovarian cancer. J. Clin. Investig. 2013, 123, 4435–4448. [Google Scholar] [CrossRef] [Green Version]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hierro, C.; Rodon, J.; Tabernero, J. Fibroblast Growth Factor (FGF) Receptor/FGF Inhibitors: Novel Targets and Strategies for Optimization of Response of Solid Tumors. Semin. Oncol. 2015, 42, 801–819. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; Gartside, M.; Powell, M.A.; Wellens, C.L.; Gao, F.; Mutch, D.G.; Goodfellow, P.J.; Pollock, P.M. FGFR2 point mutations in 466 endometrioid endometrial tumors: Relationship with MSI, KRAS, PIK3CA, CTNNB1 mutations and clinicopathological features. PLoS ONE 2012, 7, e30801. [Google Scholar] [CrossRef]
- Krook, M.A.; Reeser, J.W.; Ernst, G.; Barker, H.; Wilberding, M.; Li, G.; Chen, H.-Z.; Roychowdhury, S. Fibroblast growth factor receptors in cancer: Genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br. J. Cancer 2021, 124, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Xia, W.; Cui, Y.; Zeng, F.; Li, Y.; Yang, Z.; Hequn, C. Klotho gene polymorphisms are associated with healthy aging and longevity: Evidence from a meta-analysis. Mech. Ageing Dev. 2019, 178, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.; Li, Y.; Chen, X.; Wang, J.; Li, M.; Chen, Y.; Wu, D. FGFR-TKI resistance in cancer: Current status and perspectives. J. Hematol. Oncol. 2021, 14, 1–14. [Google Scholar] [CrossRef]
- Winterhoff, B.; Konecny, G.E. Targeting fibroblast growth factor pathways in endometrial cancer. Curr. Probl. Cancer 2017, 41, 37–47. [Google Scholar] [CrossRef]
- Uehara, Y.; Ikeda, S.; Kim, K.; Lim, H.; Adashek, J.; Persha, H.; Okamura, R.; Lee, S.; Sicklick, J.; Kato, S.; et al. Targeting the FGF/FGFR axis and its co-alteration allies. ESMO Open 2022, 7, 100647. [Google Scholar] [CrossRef]
- Nakamura, M.; Obata, T.; Daikoku, T.; Fujiwara, H. The Association and Significance of p53 in Gynecologic Cancers: The Potential of Targeted Therapy. Int. J. Mol. Sci. 2019, 20, 5482. [Google Scholar] [CrossRef] [Green Version]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. CR 2018, 37, 30. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Tahaney, W.M.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecularly targeted therapies for p53-mutant cancers. Cell. Mol. Life Sci. CMLS 2017, 74, 4171–4187. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: From molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 974. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef] [PubMed]
- Cancer Research UK. Epithelial Ovarian Cancer. Available online: https://www.cancerresearchuk.org/about-cancer/ovarian-cancer/types/epithelial-ovarian-cancers/epithelial (accessed on 22 November 2022).
- Kurman, R.J.; Shih, I.-M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [Green Version]
- Singer, G.; Oldt, R.; Cohen, Y.; Wang, B.G.; Sidransky, D.; Kurman, R.J.; Shih, I.-M. Mutations in BRAF and KRAS Characterize the Development of Low-Grade Ovarian Serous Carcinoma. JNCI J. Natl. Cancer Inst. 2003, 95, 484–486. [Google Scholar] [CrossRef] [Green Version]
- Committee on the State of the Science in Ovarian Cancer Research; Board on Health Care Services; Institute of Medicine; National Academies of Sciences, Engineering, and Medicine. Ovarian Cancers: Evolving Paradigms in Research and Care; National Academies Press (US): Washington, DC, USA, 2016; ISBN 978-0-309-38046-1.
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Prat, J. Pathology of cancers of the female genital tract. Int. J. Gynaecol. Obstet. Off. Organ Int. Fed. Gynaecol. Obstet. 2015, 131 (Suppl. 2), S132–S145. [Google Scholar] [CrossRef] [Green Version]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [Green Version]
- Samartzis, E.P.; Noske, A.; Dedes, K.J.; Fink, D.; Imesch, P. ARID1A mutations and PI3K/AKT pathway alterations in endometriosis and endometriosis-associated ovarian carcinomas. Int. J. Mol. Sci. 2013, 14, 18824–18849. [Google Scholar] [CrossRef] [Green Version]
- Zannoni, G.F.; Improta, G.; Pettinato, A.; Brunelli, C.; Troncone, G.; Scambia, G.; Fraggetta, F. Molecular status of PI3KCA, KRAS and BRAF in ovarian clear cell carcinoma: An analysis of 63 patients. J. Clin. Pathol. 2016, 69, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zeng, J.; Shen, K. PI3K/AKT/mTOR signaling pathway as a therapeutic target for ovarian cancer. Arch. Gynecol. Obstet. 2014, 290, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Li, H.; Li, W.; Gui, T.; Yang, J.; Cao, D.; Shen, K. The PI3K/AKT/mTOR pathway is a potential predictor of distinct invasive and migratory capacities in human ovarian cancer cell lines. Oncotarget 2015, 6, 25520–25532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, S.; Tsuda, H.; Takano, M.; Tamai, S.; Matsubara, O. Loss of ARID1A protein expression occurs as an early event in ovarian clear-cell carcinoma development and frequently coexists with PIK3CA mutations. Mod. Pathol. Off. J. US Can. Acad. Pathol. Inc 2012, 25, 615–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapke, N.; Chen, C.-H.; Chang, T.-C.; Chao, A.; Lu, Y.-J.; Lai, C.-H.; Tan, K.T.; Chen, H.-C.; Lu, H.-Y.; Chen, S.-J. Genetic alterations and their therapeutic implications in epithelial ovarian cancer. BMC Cancer 2021, 21, 499. [Google Scholar] [CrossRef]
- Thakur, B.; Ray, P. p53 Loses grip on PIK3CA expression leading to enhanced cell survival during platinum resistance. Mol. Oncol. 2016, 10, 1283–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilsworth, J.A.; Cochrane, D.R.; Xia, Z.; Aubert, G.; Färkkilä, A.E.M.; Horlings, H.M.; Yanagida, S.; Yang, W.; Lim, J.L.P.; Wang, Y.K.; et al. TERT promoter mutation in adult granulosa cell tumor of the ovary. Mod. Pathol. Off. J. US Can. Acad. Pathol. Inc. 2018, 31, 1107–1115. [Google Scholar] [CrossRef] [Green Version]
- Orr, B.; Edwards, R.P. Diagnosis and Treatment of Ovarian Cancer. Hematol. Oncol. Clin. North Am. 2018, 32, 943–964. [Google Scholar] [CrossRef]
- Yang, C.; Xia, B.-R.; Zhang, Z.-C.; Zhang, Y.-J.; Lou, G.; Jin, W.-L. Immunotherapy for Ovarian Cancer: Adjuvant, Combination, and Neoadjuvant. Front. Immunol. 2020, 11, 577869. [Google Scholar] [CrossRef]
- Morand, S.; Devanaboyina, M.; Staats, H.; Stanbery, L.; Nemunaitis, J. Ovarian Cancer Immunotherapy and Personalized Medicine. Int. J. Mol. Sci. 2021, 22, 6532. [Google Scholar] [CrossRef]
- Signorile, A.; De Rasmo, D.; Cormio, A.; Musicco, C.; Rossi, R.; Fortarezza, F.; Palese, L.L.; Loizzi, V.; Resta, L.; Scillitani, G.; et al. Human Ovarian Cancer Tissue Exhibits Increase of Mitochondrial Biogenesis and Cristae Remodeling. Cancers 2019, 11, 1350. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, V.W.S.; Xue, W.C.; Cheung, A.N.Y.; Ngan, H.Y.S. Association of decreased mitochondrial DNA content with ovarian cancer progression. Br. J. Cancer 2006, 95, 1087–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keserű, J.S.; Soltész, B.; Lukács, J.; Márton, É.; Szilágyi-Bónizs, M.; Penyige, A.; Póka, R.; Nagy, B. Detection of cell-free, exosomal and whole blood mitochondrial DNA copy number in plasma or whole blood of patients with serous epithelial ovarian cancer. J. Biotechnol. 2019, 298, 76–81. [Google Scholar] [CrossRef]
- Meng, X.; Schwarzenbach, H.; Yang, Y.; Müller, V.; Li, N.; Tian, D.; Shen, Y.; Gong, Z. Circulating Mitochondrial DNA is Linked to Progression and Prognosis of Epithelial Ovarian Cancer. Transl. Oncol. 2019, 12, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Zachariah, R.R.; Schmid, S.; Buerki, N.; Radpour, R.; Holzgreve, W.; Zhong, X. Levels of Circulating Cell-Free Nuclear and Mitochondrial DNA in Benign and Malignant Ovarian Tumors. Obstet. Gynecol. 2008, 112, 843–850. [Google Scholar] [CrossRef]
- Nantasupha, C.; Thonusin, C.; Charoenkwan, K.; Chattipakorn, S.; Chattipakorn, N. Metabolic reprogramming in epithelial ovarian cancer. Am. J. Transl. Res. 2021, 13, 9950–9973. [Google Scholar] [PubMed]
- Dier, U.; Shin, D.-H.; Hemachandra, L.P.M.P.; Uusitalo, L.M.; Hempel, N. Bioenergetic analysis of ovarian cancer cell lines: Profiling of histological subtypes and identification of a mitochondria-defective cell line. PLoS ONE 2014, 9, e98479. [Google Scholar] [CrossRef]
- Li, N.; Zhan, X.; Zhan, X. The lncRNA SNHG3 regulates energy metabolism of ovarian cancer by an analysis of mitochondrial proteomes. Gynecol. Oncol. 2018, 150, 343–354. [Google Scholar] [CrossRef]
- Caneba, C.A.; Bellance, N.; Yang, L.; Pabst, L.; Nagrath, D. Pyruvate uptake is increased in highly invasive ovarian cancer cells under anoikis conditions for anaplerosis, mitochondrial function, and migration. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1036–E1052. [Google Scholar] [CrossRef] [Green Version]
- Cantuaria, G.; Fagotti, A.; Ferrandina, G.; Magalhaes, A.; Nadji, M.; Angioli, R.; Penalver, M.; Mancuso, S.; Scambia, G. GLUT-1 expression in ovarian carcinoma: Association with survival and response to chemotherapy. Cancer 2001, 92, 1144–1150. [Google Scholar] [CrossRef]
- Semaan, A.; Munkarah, A.R.; Arabi, H.; Bandyopadhyay, S.; Seward, S.; Kumar, S.; Qazi, A.; Hussein, Y.; Morris, R.T.; Ali-Fehmi, R. Expression of GLUT-1 in epithelial ovarian carcinoma: Correlation with tumor cell proliferation, angiogenesis, survival and ability to predict optimal cytoreduction. Gynecol. Oncol. 2011, 121, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Lai, B.; Xiao, Y.; Pu, H.; Cao, Q.; Jing, H.; Liu, X. Overexpression of SGLT1 is correlated with tumor development and poor prognosis of ovarian carcinoma. Arch. Gynecol. Obstet. 2012, 285, 1455–1461. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Wang, J.; Zhang, L.; Wu, D.; Yu, D.; Tian, X.; Liu, J.; Jiang, X.; Shen, Y.; Zhang, L.; et al. Expressions of fatty acid synthase and HER2 are correlated with poor prognosis of ovarian cancer. Med. Oncol. 2014, 32, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Tan, Y.; Zhao, G.; Huang, K.-C.; Cardenas, H.; Matei, D.; Cheng, J.-X. Metabolic Reprogramming from Glycolysis to Fatty Acid Uptake and beta-Oxidation in Platinum-Resistant Cancer Cells. Nat. Commun. 2021, 13, 4554. [Google Scholar]
- Plewa, S.; Horała, A.; Dereziński, P.; Klupczynska, A.; Nowak-Markwitz, E.; Matysiak, J.; Kokot, Z.J. Usefulness of Amino Acid Profiling in Ovarian Cancer Screening with Special Emphasis on Their Role in Cancerogenesis. Int. J. Mol. Sci. 2017, 18, 2727. [Google Scholar] [CrossRef] [Green Version]
- Tanwar, D.K.; Parker, D.J.; Gupta, P.; Spurlock, B.; Alvarez, R.D.; Basu, M.K.; Mitra, K. Crosstalk between the mitochondrial fission protein, Drp1, and the cell cycle is identified across various cancer types and can impact survival of epithelial ovarian cancer patients. Oncotarget 2016, 7, 60021–60037. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Kim, B.; Cho, U.; Park, I.S.; Kim, S.I.; Dhanasekaran, D.N.; Tsang, B.K.; Song, Y.S. Mitochondrial fission causes cisplatin resistance under hypoxic conditions via ROS in ovarian cancer cells. Oncogene 2019, 38, 7089–7105. [Google Scholar] [CrossRef]
- Kong, B.; Wang, Q.; Fung, E.; Xue, K.; Tsang, B.K. p53 Is Required for Cisplatin-induced Processing of the Mitochondrial Fusion Protein L-Opa1 That Is Mediated by the Mitochondrial Metallopeptidase Oma1 in Gynecologic Cancers. J. Biol. Chem. 2014, 289, 27134–27145. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Mao, Y.; Xi, S.; Wang, X.; Sun, L. Nutrient Starvation Sensitizes Human Ovarian Cancer SKOV3 Cells to BH3 Mimetic via Modulation of Mitochondrial Dynamics. Anat. Rec. 2017, 300, 326–339. [Google Scholar] [CrossRef]
- Kingnate, C.; Charoenkwan, K.; Kumfu, S.; Chattipakorn, N.; Chattipakorn, S.C. Possible Roles of Mitochondrial Dynamics and the Effects of Pharmacological Interventions in Chemoresistant Ovarian Cancer. EBioMedicine 2018, 34, 256–266. [Google Scholar] [CrossRef]
- Ong, S.-B.; Gustafsson, A.B. New roles for mitochondria in cell death in the reperfused myocardium. Cardiovasc. Res. 2012, 94, 190–196. [Google Scholar] [CrossRef] [Green Version]
- Wappler, E.A.; Institoris, A.; Dutta, S.; Katakam, P.V.G.; Busija, D.W. Mitochondrial dynamics associated with oxygen-glucose deprivation in rat primary neuronal cultures. PLoS ONE 2013, 8, e63206. [Google Scholar] [CrossRef] [PubMed]
- Ferri-Borgogno, S.; Barui, S.; McGee, A.M.; Griffiths, T.; Singh, P.K.; Piett, C.G.; Ghosh, B.; Bhattacharyya, S.; Singhi, A.; Pradhan, K.; et al. Paradoxical Role of AT-rich Interactive Domain 1A in Restraining Pancreatic Carcinogenesis. Cancers 2020, 12, 2695. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.N.; Roberts, C.W.M. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfert, A.; Moreno, N.; Kerl, K. The BAF complex in development and disease. Epigenetics Chromatin 2019, 12, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Haswell, J.R.; Roberts, C.W.M. Molecular pathways: SWI/SNF (BAF) complexes are frequently mutated in cancer--mechanisms and potential therapeutic insights. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalimuthu, S.N.; Chetty, R. Gene of the month: SMARCB1. J. Clin. Pathol. 2016, 69, 484–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, E.; Liu, B.; Li, C.; Liu, T.; Rutter, J.; Grossman, D. Loss of p16INK4A stimulates aberrant mitochondrial biogenesis through a CDK4/Rb-independent pathway. Oncotarget 2017, 8, 55848. [Google Scholar] [CrossRef] [Green Version]
- Luo, Q.; Wu, X.; Liu, Z. Remodeling of the ARID1A tumor suppressor. Cancer Lett. 2020, 491, 26. [Google Scholar] [CrossRef]
- Trost, T.; Haines, J.; Dillon, A.; Mersman, B.; Robbins, M.; Thomas, P.; Hubert, A. Characterizing the role of SWI/SNF-related chromatin remodeling complexes in planarian regeneration and stem cell function. Stem Cell Res. 2018, 32, 91–103. [Google Scholar] [CrossRef]
- Varga, J.; Kube, M.; Luck, K.; Schick, S. The BAF chromatin remodeling complexes: Structure, function, and synthetic lethalities. Biochem. Soc. Trans. 2021, 49, 1489–1503. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.W.; Hong, A.L. SMARCB1-Deficient Cancers: Novel Molecular Insights and Therapeutic Vulnerabilities. Cancers 2022, 14, 3645. [Google Scholar] [CrossRef]
- Kohashi, K.; Oda, Y. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Sci. 2017, 108, 547–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, J.; Mandal, P.; Wang, T.-L.; Shih, I.-M. Treating ARID1A mutated cancers by harnessing synthetic lethality and DNA damage response. J. Biomed. Sci. 2022, 29, 71. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R. ARID1A loss in cancer: Towards a mechanistic understanding. Pharmacol. Ther. 2018, 190, 15–23. [Google Scholar] [CrossRef]
- Alldredge, J.K.; Eskander, R.N. EZH2 inhibition in ARID1A mutated clear cell and endometrioid ovarian and endometrioid endometrial cancers. Gynecol. Oncol. Res. Pract. 2017, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garczyk, S.; Schneider, U.; Lurje, I.; Becker, K.; Vögeli, T.A.; Gaisa, N.T.; Knüchel, R. ARID1A-deficiency in urothelial bladder cancer: No predictive biomarker for EZH2-inhibitor treatment response? PLoS ONE 2018, 13, e0202965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornen, S.; Adelaide, J.; Bertucci, F.; Finetti, P.; Guille, A.; Birnbaum, D.J.; Birnbaum, D.; Chaffanet, M. Mutations and deletions of ARID1A in breast tumors. Oncogene 2012, 31, 4255–4256. [Google Scholar] [CrossRef] [Green Version]
- Guan, B.; Wang, T.-L.; Shih, I.-M. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011, 71, 6718–6727. [Google Scholar] [CrossRef] [Green Version]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell 2019, 35, 177–190.e8. [Google Scholar] [CrossRef] [Green Version]
- Odnokoz, O.; Wavelet-Vermuse, C.; Hophan, S.L.; Bulun, S.; Wan, Y. ARID1 proteins: From transcriptional and post-translational regulation to carcinogenesis and potential therapeutics. Epigenomics 2021, 13, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Tang, C. The Role of ARID1A in Tumors: Tumor Initiation or Tumor Suppression? Front. Oncol. 2021, 11, 745187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shetty, M.; Clemente, V.; Linder, S.; Bazzaro, M. Targeting Mitochondrial Metabolism in Clear Cell Carcinoma of the Ovaries. Int. J. Mol. Sci. 2021, 22, 4750. [Google Scholar] [CrossRef]
- Lissanu Deribe, Y.; Sun, Y.; Terranova, C.; Khan, F.; Martinez-Ledesma, J.; Gay, J.; Gao, G.; Mullinax, R.A.; Khor, T.; Feng, N.; et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat. Med. 2018, 24, 1047–1057. [Google Scholar] [CrossRef]
- Liu, X.; Li, Z.; Wang, Z.; Liu, F.; Zhang, L.; Ke, J.; Xu, X.; Zhang, Y.; Yuan, Y.; Wei, T.; et al. Chromatin Remodeling Induced by ARID1A Loss in Lung Cancer Promotes Glycolysis and Confers JQ1 Vulnerability. Cancer Res. 2022, 82, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Jing, J.; Yuan, Y.; Feng, J.; Han, X.; Chen, S.; Li, X.; Peng, W.; Xu, J.; Ho, T.-V.; et al. Arid1a-Plagl1-Hh signaling is indispensable for differentiation-associated cell cycle arrest of tooth root progenitors. Cell Rep. 2021, 35, 108964. [Google Scholar] [CrossRef] [PubMed]
- Suryo Rahmanto, Y.; Jung, J.-G.; Wu, R.-C.; Kobayashi, Y.; Heaphy, C.M.; Meeker, A.K.; Wang, T.-L.; Shih, I.-M. Inactivating ARID1A Tumor Suppressor Enhances TERT Transcription and Maintains Telomere Length in Cancer Cells. J. Biol. Chem. 2016, 291, 9690–9699. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Friedland, S.C.; Alexander, W.; Myers, J.A.; Wang, W.; O’Dell, M.R.; Getman, M.; Whitney-Miller, C.L.; Agostini-Vulaj, D.; Huber, A.R.; et al. Arid1a mutation suppresses TGF-β signaling and induces cholangiocarcinoma. Cell Rep. 2022, 40, 111253. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Tu, W.; Yang, L.; Peng, G.; Yang, L. ARID1A deficiency and immune checkpoint blockade therapy: From mechanisms to clinical application. Cancer Lett. 2020, 473, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W.M. Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet. 2014, 207, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Munro, J.; Barr, N.I.; Ireland, H.; Morrison, V.; Parkinson, E.K. Histone deacetylase inhibitors induce a senescence-like state in human cells by a p16-dependent mechanism that is independent of a mitotic clock. Exp. Cell Res. 2004, 295, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Bitler, B.G.; Wu, S.; Park, P.H.; Hai, Y.; Aird, K.M.; Wang, Y.; Zhai, Y.; Kossenkov, A.V.; Vara-Ailor, A.; Rauscher, F.J.; et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 2017, 19, 962–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, S.; Rao, S.V.; Sutton, J.; Cheeseman, D.; Dunn, S.; Papachristou, E.K.; Prada, J.-E.G.; Couturier, D.-L.; Kumar, S.; Kishore, K.; et al. ARID1A influences HDAC1/BRD4 activity, intrinsic proliferative capacity and breast cancer treatment response. Nat. Genet. 2020, 52, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, T.; Park, P.H.; Wu, S.; Fatkhutdinov, N.; Karakashev, S.; Nacarelli, T.; Kossenkov, A.V.; Speicher, D.W.; Jean, S.; Zhang, L.; et al. Repurposing Pan-HDAC Inhibitors for ARID1A-Mutated Ovarian Cancer. Cell Rep. 2018, 22, 3393–3400. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Torrini, C.; Zhao, J.; Bianchetti, E.; Mela, A.; Humala, N.; Mahajan, A.; et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J. Clin. Investig. 2020, 130, 3699–3716. [Google Scholar] [CrossRef]
- Chang, P.; Li, H.; Hu, H.; Li, Y.; Wang, T. The Role of HDAC6 in Autophagy and NLRP3 Inflammasome. Front. Immunol. 2021, 12, 763831. [Google Scholar] [CrossRef]
- Ma, C.; Hu, K.; Ullah, I.; Zheng, Q.-K.; Zhang, N.; Sun, Z.-G. Molecular Mechanisms Involving the Sonic Hedgehog Pathway in Lung Cancer Therapy: Recent Advances. Front. Oncol. 2022, 12, 729088. [Google Scholar] [CrossRef]
- Jeng, K.-S.; Chang, C.-F.; Lin, S.-S. Sonic Hedgehog Signaling in Organogenesis, Tumors, and Tumor Microenvironments. Int. J. Mol. Sci. 2020, 21, 758. [Google Scholar] [CrossRef] [Green Version]
- Haybaeck, J.; Taucher, V. Mechanisms of molecular carcinogenesis; Springer: Cham, UK, 2017; Volume 2, ISBN 978-3-319-53661-3. [Google Scholar]
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; de Sampaio e Spohr, T.C.L. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef]
- Andrianantoandro, E. Hedgehog Drives Glucose Metabolism. Sci. Signal. 2012, 5, 3715. [Google Scholar] [CrossRef]
- Teperino, R.; Amann, S.; Bayer, M.; McGee, S.L.; Loipetzberger, A.; Connor, T.; Jaeger, C.; Kammerer, B.; Winter, L.; Wiche, G.; et al. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell 2012, 151, 414–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirou, L.; Russo, M.; Faure, H.; Pellegrino, G.; Demongin, C.; Daynac, M.; Sharif, A.; Amosse, J.; Le Lay, S.; Denis, R.; et al. Sonic Hedgehog receptor Patched deficiency in astrocytes enhances glucose metabolism in mice. Mol. Metab. 2021, 47, 101172. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, J.B.; Popli, P.; Sankhwar, P.; Shukla, V.; Dwivedi, A. Sonic hedgehog protects endometrial hyperplasial cells against oxidative stress via suppressing mitochondrial fission protein dynamin-like GTPase (Drp1). Free Radic. Biol. Med. 2018, 129, 582–599. [Google Scholar] [CrossRef]
- Zhan, X.; Shi, X.; Zhang, Z.; Chen, Y.; Wu, J.I. Dual role of Brg chromatin remodeling factor in Sonic hedgehog signaling during neural development. Proc. Natl. Acad. Sci. USA 2011, 108, 12758–12763. [Google Scholar] [CrossRef] [Green Version]
- Buj, R.; Aird, K.M. p16: Cycling off the beaten path. Mol. Cell. Oncol. 2019, 6, e1677140. [Google Scholar] [CrossRef] [Green Version]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilgendorf, K.I.; Leshchiner, E.S.; Nedelcu, S.; Maynard, M.A.; Calo, E.; Ianari, A.; Walensky, L.D.; Lees, J.A. The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev. 2013, 27, 1003–1015. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, N.C.; Liu, T.; Cassidy, P.; Leachman, S.A.; Boucher, K.M.; Goodson, A.G.; Samadashwily, G.; Grossman, D. The p16INK4A tumor suppressor regulates cellular oxidative stress. Oncogene 2011, 30, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.-Y.; Zhu, S.-S.; Chen, X.-J.; Zhu, J.; Chen, Q.; Zhang, Y.-Q.; Zhang, C.-L.; Guo, T.-T.; Zhang, L.-M. ARID1A suppresses malignant transformation of human pancreatic cells via mediating senescence-associated miR-503/CDKN2A regulatory axis. Biochem. Biophys. Res. Commun. 2017, 493, 1018–1025. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, C.; Yu, S.; Jia, C.; Yan, J.; Lu, Z.; Chen, J. Loss of ARID1A Expression Correlates with Tumor Differentiation and Tumor Progression Stage in Pancreatic Ductal Adenocarcinoma. Technol. Cancer Res. Treat. 2018, 17, 1533034618754475. [Google Scholar] [CrossRef] [Green Version]
- Icreverzi, A.; de la Cruz, A.F.; Van Voorhies, W.A.; Edgar, B.A. Drosophila cyclin D/Cdk4 regulates mitochondrial biogenesis and aging and sensitizes animals to hypoxic stress. Cell Cycle Georget. Tex 2012, 11, 554–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riou, R.; Ladli, M.; Gerbal-Chaloin, S.; Bossard, P.; Gougelet, A.; Godard, C.; Loesch, R.; Lagoutte, I.; Lager, F.; Calderaro, J.; et al. ARID1A loss in adult hepatocytes activates β-catenin-mediated erythropoietin transcription. eLife 2020, 9, e53550. [Google Scholar] [CrossRef]
- Reske, J.J.; Wilson, M.R.; Holladay, J.; Siwicki, R.A.; Skalski, H.; Harkins, S.; Adams, M.; Risinger, J.I.; Hostetter, G.; Lin, K.; et al. Co-existing TP53 and ARID1A mutations promote aggressive endometrial tumorigenesis. PLoS Genet. 2021, 17, e1009986. [Google Scholar] [CrossRef] [PubMed]
- Huynh, L.K.; Hipolito, C.J.; Ten Dijke, P. A Perspective on the Development of TGF-β Inhibitors for Cancer Treatment. Biomolecules 2019, 9, 743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Chen, Y.-G. The Interplay Between TGF-β Signaling and Cell Metabolism. Front. Cell Dev. Biol. 2022, 10, 846723. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, M.; Xiong, L.; Fan, J.; Zhou, Y.; Li, H.; Peng, X.; Zhong, Z.; Wang, Y.; Huang, F.; et al. Drp1-mediated mitochondrial fission promotes renal fibroblast activation and fibrogenesis. Cell Death Dis. 2020, 11, 29. [Google Scholar] [CrossRef] [Green Version]
- Casalena, G.; Daehn, I.; Bottinger, E. Transforming growth factor-β, bioenergetics, and mitochondria in renal disease. Semin. Nephrol. 2012, 32, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Huang, J.; Wang, G. Mitochondria, Telomeres and Telomerase Subunits. Front. Cell Dev. Biol. 2019, 7, 274. [Google Scholar] [CrossRef] [Green Version]
- Dratwa, M.; Wysoczańska, B.; Łacina, P.; Kubik, T.; Bogunia-Kubik, K. TERT—Regulation and Roles in Cancer Formation. Front. Immunol. 2020, 11, 589929. [Google Scholar] [CrossRef]
- Colebatch, A.J.; Dobrovic, A.; Cooper, W.A. TERT gene: Its function and dysregulation in cancer. J. Clin. Pathol. 2019, 72, 281–284. [Google Scholar] [CrossRef]
- Ebata, H.; Shima, T.; Iizuka, R.; Uemura, S. Accumulation of TERT in Mitochondria Shows Two Opposing Effects on Apoptosis. bioRxiv 2022. [Google Scholar] [CrossRef]
- Santos, J.H.; Meyer, J.N.; Skorvaga, M.; Annab, L.A.; Van Houten, B. Mitochondrial hTERT exacerbates free-radical-mediated mtDNA damage. Aging Cell 2004, 3, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haendeler, J.; Dröse, S.; Büchner, N.; Jakob, S.; Altschmied, J.; Goy, C.; Spyridopoulos, I.; Zeiher, A.M.; Brandt, U.; Dimmeler, S. Mitochondrial Telomerase Reverse Transcriptase Binds to and Protects Mitochondrial DNA and Function from Damage. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Lamb, R.; Ozsvari, B.; Bonuccelli, G.; Smith, D.L.; Pestell, R.G.; Martinez-Outschoorn, U.E.; Clarke, R.B.; Sotgia, F.; Lisanti, M.P. Dissecting tumor metabolic heterogeneity: Telomerase and large cell size metabolically define a sub-population of stem-like, mitochondrial-rich, cancer cells. Oncotarget 2015, 6, 21892–21905. [Google Scholar] [CrossRef] [Green Version]
- Viswanath, P.; Batsios, G.; Ayyappan, V.; Taglang, C.; Gillespie, A.M.; Larson, P.E.Z.; Luchman, H.A.; Costello, J.F.; Pieper, R.O.; Ronen, S.M. Metabolic imaging detects elevated glucose flux through the pentose phosphate pathway associated with TERT expression in low-grade gliomas. Neuro-Oncol. 2021, 23, 1509–1522. [Google Scholar] [CrossRef]
- Green, P.D.; Sharma, N.K.; Santos, J.H. Telomerase Impinges on the Cellular Response to Oxidative Stress Through Mitochondrial ROS-Mediated Regulation of Autophagy. Int. J. Mol. Sci. 2019, 20, 1509. [Google Scholar] [CrossRef] [Green Version]
- Guterres, A.N.; Villanueva, J. Targeting telomerase for cancer therapy. Oncogene 2020, 39, 5811–5824. [Google Scholar] [CrossRef]
- Hemann, M.T.; Strong, M.A.; Hao, L.Y.; Greider, C.W. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 2001, 107, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Klinge, C.M. Estrogenic control of mitochondrial function and biogenesis. J. Cell. Biochem. 2008, 105, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Wappler, E.A.; Gál, A.; Skopál, J.; Nagy, Z. Single, high-dose 17β-estradiol therapy has anti-apoptotic effect and induces cerebral plasticity following transient forebrain ischemia in gerbils (Short communication). Acta Physiol. Hung. 2011, 98, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Wappler, E.A.; Felszeghy, K.; Varshney, M.; Mehra, R.D.; Nyakas, C.; Nagy, Z. Brain Plasticity Following Ischemia: Effect of Estrogen and Other Cerebroprotective Drugs. In Advances in the Treatment of Ischemic Stroke; Balestrino, M., Ed.; InTech: London, UK, 2012; ISBN 978-953-51-0136-9. [Google Scholar]
- Sastre-Serra, J.; Nadal-Serrano, M.; Pons, D.G.; Roca, P.; Oliver, J. Mitochondrial dynamics is affected by 17β-estradiol in the MCF-7 breast cancer cell line. Effects on fusion and fission related genes. Int. J. Biochem. Cell Biol. 2012, 44, 1901–1905. [Google Scholar] [CrossRef] [PubMed]
- Mattingly, K.A.; Ivanova, M.M.; Riggs, K.A.; Wickramasinghe, N.S.; Barch, M.J.; Klinge, C.M. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol. Endocrinol. Baltim. Md. 2008, 22, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Klinge, C.M. Estrogenic control of mitochondrial function. Redox Biol. 2020, 31, 101435. [Google Scholar] [CrossRef]
- Shiba, S.; Ikeda, K.; Horie-Inoue, K.; Nakayama, A.; Tanaka, T.; Inoue, S. Deficiency of COX7RP, a mitochondrial supercomplex assembly promoting factor, lowers blood glucose level in mice. Sci. Rep. 2017, 7, 7606. [Google Scholar] [CrossRef] [PubMed] [Green Version]






| Subtype | Clinicopathological Definition |
|---|---|
| Luminal A | “Luminal A like” ER-positive HER2-negative Ki67 low PR high |
| Luminal B | “Luminal B-like (HER2-negative)” ER-positive HER2-negative and either Ki67 high or PR low “Luminal B-like (HER2-positive)” ER-positive HER2-positive Any Ki67 Any PR |
| HER2 positive | “HER2-positive (non-luminal)” HER2-positive ER and PR absent |
| Triple negative | “Triple-negative” ER and PR absent HER2-negative |
| Metabolic Pathway | Enzyme/Protein | Role in Breast Cancer |
|---|---|---|
| Anaerobic glycolysis | Enolases | α-Enolase gene expression correlates with tumor size and shorter disease-free interval [25,26] |
| Pyruvate kinase | Levels elevated in aggressive breast cancer type [28] | |
| Oxidative phosphorylation (OXPHOS) | Complex I, II, III, and IV | Aggressive breast cancer shows the broadest OXPHOS defect in cell lines [29] |
| UQCRFS1 and UQCRH (complex III subunits) | Increased expression in breast tumors compared to normal breast tissue [29] | |
| Other metabolic markers | Hydratases | MTHFD2 protein content 3-fold decreased in breast cancer cell line [31] |
| Dehydrogenases | IDH2 expression elevated in breast cancer cell lines. Expression is positively associated with overall survival [32]. | |
| Oxidases | Coproporphyrinogen III oxidase expression elevated in Adriamycin-resistant breast cancer cell lines [33] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czegle, I.; Huang, C.; Soria, P.G.; Purkiss, D.W.; Shields, A.; Wappler-Guzzetta, E.A. The Role of Genetic Mutations in Mitochondrial-Driven Cancer Growth in Selected Tumors: Breast and Gynecological Malignancies. Life 2023, 13, 996. https://doi.org/10.3390/life13040996
Czegle I, Huang C, Soria PG, Purkiss DW, Shields A, Wappler-Guzzetta EA. The Role of Genetic Mutations in Mitochondrial-Driven Cancer Growth in Selected Tumors: Breast and Gynecological Malignancies. Life. 2023; 13(4):996. https://doi.org/10.3390/life13040996
Chicago/Turabian StyleCzegle, Ibolya, Chelsea Huang, Priscilla Geraldine Soria, Dylan Wesley Purkiss, Andrea Shields, and Edina Amalia Wappler-Guzzetta. 2023. "The Role of Genetic Mutations in Mitochondrial-Driven Cancer Growth in Selected Tumors: Breast and Gynecological Malignancies" Life 13, no. 4: 996. https://doi.org/10.3390/life13040996
APA StyleCzegle, I., Huang, C., Soria, P. G., Purkiss, D. W., Shields, A., & Wappler-Guzzetta, E. A. (2023). The Role of Genetic Mutations in Mitochondrial-Driven Cancer Growth in Selected Tumors: Breast and Gynecological Malignancies. Life, 13(4), 996. https://doi.org/10.3390/life13040996