


Managing Anemia: Point of Convergence for Heart Failure and Chronic Kidney Disease?



 , ,
, ,
Abstract
:1. Introduction
2. Epidemiology
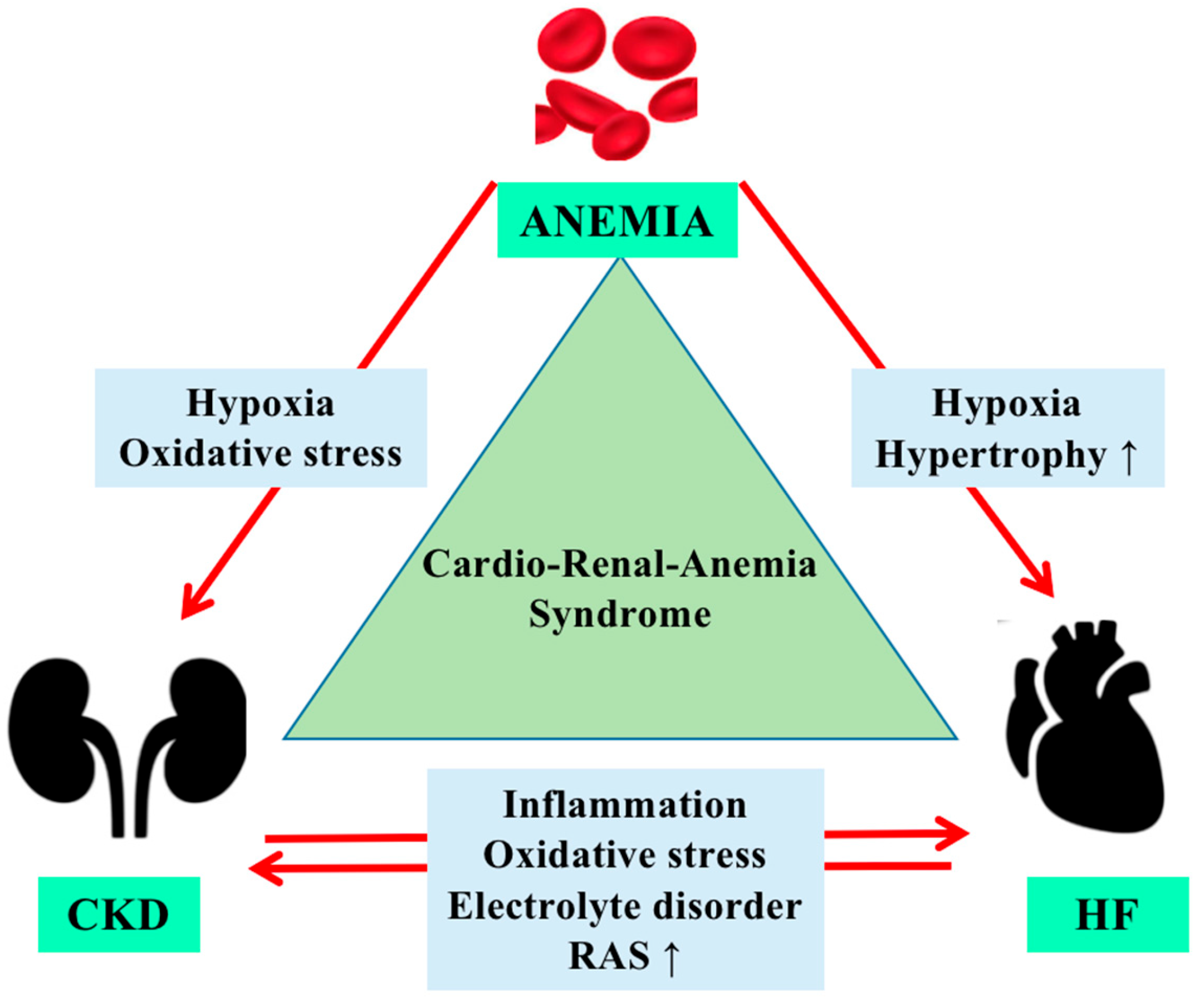
3. Physiopathology of Anemia in Cardiorenal Syndrome
4. Managing Anemia in Cardiorenal Patients
4.1. Intravenous Iron
4.2. Erythropoiesis-Stimulating Agents
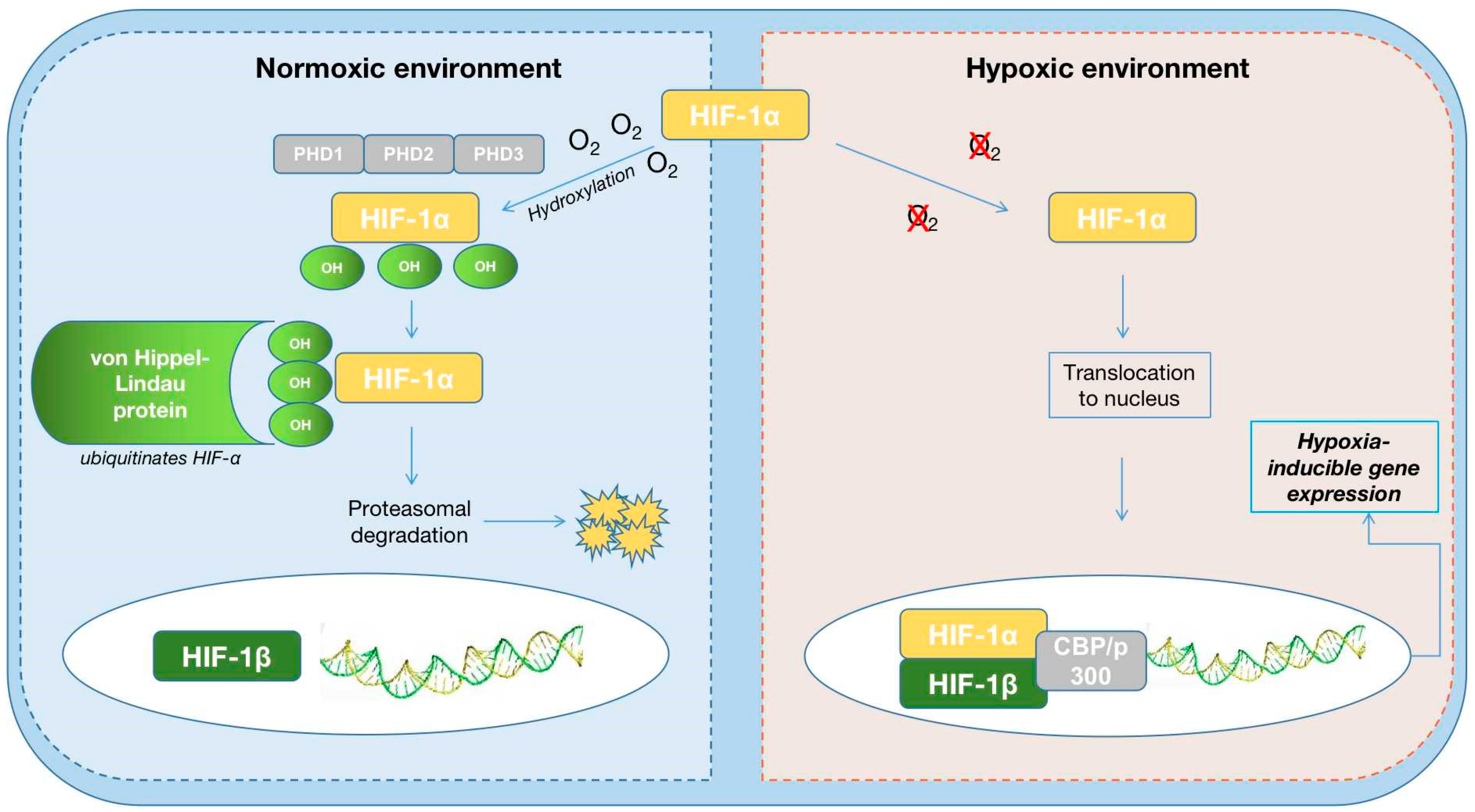
4.3. HIF-PH Inhibitors

{kind=link}
{kind=link}
| Roxadustat | Daprodustat | Vadadustat | Molidustat | Enarodustat | Desidustat | |
|---|---|---|---|---|---|---|
| PHD target | all 3 HIF-PHDs | inhibits PHD1 and PHD3 | all three PHDs | mainly inhibits PHD2 | all 3 HIF-PHDs | |
| Benefic Effects |
|
|
|
|
| increased EPO levels and decreased hepcidin and low-density lipoprotein cholesterol (LDL-C) levels, improves EPO-sensitivity by decreasing IL-6, IL-1β, and anti-EPO antibodies [87] |
| Adverse effects |
|
|
|
|
| Pyrexia, vomiting, asthenia, peripheral oedema |
| Half life (h) | 12~15 h | 1.3~2.5 h | 7~9 h | 4~10 h | 15 h | 6–15 h |
| Study population | DD (HD/DP) and NDD | DD (HD/DP) and NDD patients with HF and renal anemia | DD (HD/DP) and NDD | DD (HD/DP) and NDD | DD (HD/DP) and NDD | treatment of anemia associated with CKD (DD and NDD), COVID-2019 infections and chemotherapy induced anemia |
4.4. Hepcidin Antagonist
| Inflammatory status | Hepcidin antagonists anti-IL-6 receptor antibody [91,95] |
| Iron deficiency | Intravenous iron therapy [38,42,43] |
| Hypoxic environment | Erythropoiesis-stimulating agents HIF-PH inhibitors SGLT2 [58,67,88] |
5. Future Perspectives
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rangaswami, J.; Bhalla, V.; Blair, J.E.; Chang, T.I.; Costa, S.; Lentine, K.L.; Lerma, E.V.; Mezue, K.; Molitch, M.; Mullens, W.; et al. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement From the American Heart Association. Circulation 2019, 139, e840–e878. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Costanzo, M.R.; Bellomo, R.; Maisel, A.S. Cardiorenal syndromes: Definition and classification. Contrib. Nephrol. 2010, 164, 33–38. [Google Scholar] [PubMed]
- Shi, S.; Zhang, B.; Li, Y.; Xu, X.; Lv, J.; Jia, Q.; Chai, R.; Xue, W.; Li, Y.; Wang, Y.; et al. Mitochondrial Dysfunction: An Emerging Link in the Pathophysiology of Cardiorenal Syndrome. Front. Cardiovasc. Med. 2022, 9, 837270. [Google Scholar] [CrossRef] [PubMed]
- Rivera, R.F.; Alibrandi, M.T.S.; Di Lullo, L. The Cardiorenal Anemia Syndrome. Part One: Epidemiology and Clinical Aspects. G. Tec. Nefrol. Dial. 2017, 29, 196–202. [Google Scholar]
- Farmakis, D.; Filippatos, G. Cardiorenal-anemia syndrome—Definition, epidemiology and management: The Cardiologist’s view. Hippokratia 2011, 15 (Suppl. 2), 9–14. [Google Scholar]
- McLean, E.; Cogswell, M.; Egli, I.; Wojdyla, D.; De Benoist, B. Worldwide prevalence of anaemia, WHO Vitamin and Mineral Nutrition Information System, 1993–2005. Public Health Nutr. 2009, 12, 444–454. [Google Scholar] [CrossRef]
- Anker, S.D.; Comin Colet, J.; Filippatos, G.; Willenheimer, R.; Dickstein, K.; Drexler, H.; Lüscher, T.M.; Bart, B.; Banasiak, W.; Niegowska, J.; et al. FAIR-HF Trial Investigators. Ferric carboxymaltose in patients with heart failure and iron deficiency. N. Engl. J. Med. 2009, 361, 2436–2448. [Google Scholar] [CrossRef]
- Gutiérrez, O.M. Treatment of Iron Deficiency Anemia in CKD and End-Stage Kidney Disease. Kidney Int. Rep. 2021, 6, 2261–2269. [Google Scholar] [CrossRef]
- Kumar, U.; Wettersten, N.; Garimella, P.S. Cardiorenal Syndrome: Pathophysiology. Cardiol. Clin. 2019, 37, 251–265. [Google Scholar] [CrossRef]
- McCullough, P.A. Anemia of cardiorenal syndrome. Kidney Int. 2021, 11, 35–45. [Google Scholar] [CrossRef]
- Portolés, J.; Martín, L.; Broseta, J.J.; Cases, A. Anemia in Chronic Kidney Disease: From Pathophysiology and Current Treatments, to Future Agents. Front. Med. 2021, 8, 642296. [Google Scholar] [CrossRef] [PubMed]
- Chertow, G.M.; Pergola, P.E.; Farag, Y.M.; Agarwal, R.; Arnold, S.; Bako, G.; Block, G.A.; Burke, S.; Castillo, F.P.; Jardine, A.G.; et al. Vadadustat in Patients with Anemia and Non–Dialysis-Dependent CKD. N. Engl. J. Med. 2021, 384, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- KDIGO Anemia Working Group. KDIGO clinical practice guideline for anemia in chronic kidney disease. Kidney Int. 2012, 2, 279–335. [Google Scholar]
- Van der Weerd, N.C.; Grooteman, M.P.; Nubé, M.J.; Ter Wee, P.M.; Swinkels, D.W.; Gaillard, C.A. Hepcidin in chronic kidney disease: Not an anaemia management tool, but promising as a cardiovascular biomarker. Neth. J. Med. 2015, 73, 108–118. [Google Scholar] [PubMed]
- Deray, G.; Heurtier, A.; Grimaldi, A. Anemia and diabetes. Am. J. Nephrol. 2004, 24, 522–526. [Google Scholar] [CrossRef]
- Besarab, A.; Hörl, W.H.; Silverberg, D. Iron Metabolism, Iron Deficiency, Thrombocytosis, and the Cardiorenal Anemia Syn-drome. Oncologist 2009, 14, 22–33. [Google Scholar] [CrossRef]
- Marecos, C.; Falcao, L.M. Anemia and cardiorenal syndrome in heart failure: Review article. Med. Interna 2010, 17, 236–245. [Google Scholar]
- Kuriyama, S.; Maruyama, Y.; Honda, H. A new insight into the treatment of renal anemia with HIF stabilizer. Ren. Replace Therapy 2020, 6, 63. [Google Scholar] [CrossRef]
- Silverberg, D.S.; Wexler, D.; Iaina, A.; Schwartz, D. Correction of Iron Deficiency in the Cardiorenal Syndrome. Int. J. Nephrol. 2011, 2011, 365301. [Google Scholar] [CrossRef]
- Carrilho, P. Intravenous iron in heart failure and chronic kidney disease. Nefrologia 2021, 41, 403–411. [Google Scholar] [CrossRef]
- Honda, H.; Kobayashi, Y.; Onuma, S.; Shibagaki, K.; Yuza, T.; Hirao, K.; Yamamoto, T.; Tomosugi, N.; Shibata, T. Associations among Erythroferrone and Biomarkers of Erythropoiesis and Iron Metabolism, and Treatment with Long-Term Erythropoiesis-Stimulating Agents in Patients on Hemodialysis. PLoS ONE 2016, 11, e0151601. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Erythropoietic regulators of iron metabolism. Free Radic. Biol. Med. 2018, 133, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Kazory, A.; Ross, E.A. Anemia: The Point of Convergence or Divergence for Kidney Disease and Heart Failure? J. Am. Coll. Cardiol. 2009, 53, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, W.M.; Wiesener, M.S.; Scigalla, P.; Chou, J.; Schmieder, R.E.; Günzler, V.; Eckardt, K.-U. Inhibition of Prolyl Hydroxylases Increases Erythropoietin Production in ESRD. J. Am. Soc. Nephrol. 2010, 21, 2151–2156. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Gupta, N.; Wish, J.B. Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors: A Potential New Treatment for Anemia in Patients with CKD. Am. J. Kidney Dis. 2017, 69, 815–826. [Google Scholar] [CrossRef]
- Caramelo, C.; Justo, S.; Gil, P. Anemia in heart failure: Pathophysiology, pathogenesis, treatment, and incognitae. Rev. Esp. Cardiol. 2007, 60, 848–860. [Google Scholar] [CrossRef]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, N.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1 in hypoxia- mediated apoptosis, cell proliferation and Tumor angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef]
- Packer, M. Mutual Antagonism of Hypoxia-Inducible Factor Isoforms in Cardiac, Vascular, and Renal Disorders. JACC Basic Transl. Sci. 2020, 5, 961–968. [Google Scholar] [CrossRef]
- Jelkmann, W. Molecular Biology of Erythropoietin. Intern. Med. 2004, 43, 649–659. [Google Scholar] [CrossRef]
- Eckardt, K. Anaemia in end-stage renal disease: Pathophysiological considerations. Nephrol. Dial. Transplant. 2001, 16, 2–8. [Google Scholar] [CrossRef] [PubMed]
- West, J.B. Physiological effects of chronic hypoxia. N. Engl. J. Med. 2017, 376, 1965–1971. [Google Scholar] [CrossRef]
- Eckardt, K.-U. The noblesse of kidney physiology. Kidney Int. 2019, 96, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Miao, M.; Wu, M.; Li, Y.; Zhang, L.; Jin, Q.; Fan, J.; Xu, X.; Gu, R.; Hao, H.; Zhang, A.; et al. Clinical Potential of Hypoxia Inducible Factors Prolyl Hydroxylase Inhibitors in Treating Nonanemic Diseases. Front. Pharmacol. 2022, 13, 837249. [Google Scholar] [CrossRef]
- Wang, B.; Li, Z.-L.; Zhang, Y.-L.; Wen, Y.; Gao, Y.-M.; Liu, B.-C. Hypoxia and chronic kidney disease. EBioMedicine 2022, 77, 103942. [Google Scholar] [CrossRef]
- Singer, C.E.; Vasile, C.M.; Popescu, M.; Popescu, A.I.S.; Marginean, I.C.; Iacob, G.A.; Popescu, M.D.; Marginean, C.M. Role of Iron Defificiency in Heart Failure—Clinical and Treatment Approach: An Overview. Diagnostics 2023, 13, 304. [Google Scholar] [CrossRef]
- Guedes, M.; Robinson, B.M.; Obrador, G.; Tong, A.; Pisoni, R.L.; Pecoits-Filho, R. Management of Anemia in Nondialysis Chronic Kidney Disease: Current Recommendations, Real-World Practice, and Patient Perspectives. Kidney360 2020, 1, 855–862. [Google Scholar] [CrossRef]
- Okonko, D.O.; Grzeslo, A.; Witkowski, T.; Mandal, A.K.; Slater, R.M.; Roughton, M.; Foldes, G.; Thum, T.; Majda, J.; Banasiak, W.; et al. Effect of intravenous iron sucrose on exercise tolerance in anemic and non-anemic patients with symptomatic chronic heart failure and iron deficiency FERRIC-HF: A randomized, controlled, observer-blinded trial. J. Am. Coll. Cardiol. 2008, 51, 103–112. [Google Scholar] [CrossRef]
- Kortman, G.A.M.; Reijnders, D.; Swinkels, D.W. Oral iron supplementation: Potential implications for the gut microbiome and metabolome in patients with CKD. Hemodial. Int. 2017, 21, S28–S36. [Google Scholar] [CrossRef]
- Cigarran Guldris, S.; Gonzalez Parra, E.; Cases Amenos, A. Gut microbiota in chronic kidney disease. Nefrologia 2017, 37, 9–19. [Google Scholar] [CrossRef]
- Ben-Assa, E.; Shacham, Y.; Shashar, M.; Leshem-Rubinow, E.; Gal-Oz, A.; Schwartz, I.F.; Schwartz, D.; Silverberg, D.S.; Chernin, G. Target hemoglobin may be achieved with intravenous iron alone in anemic patients with cardiorenal syndrome: An observational study. Cardiorenal Med. 2015, 5, 246–253. [Google Scholar] [CrossRef]
- Gupta, A.; Lin, V.; Guss, C.; Pratt, R.; Ikizler, T.A.; Besarab, A. Ferric pyrophosphate citrate administered via dialysate reduces erythropoiesis-stimulating agent use and maintains hemoglobin in hemodialysis patients. Kidney Int. 2015, 88, 1187–1194. [Google Scholar] [CrossRef]
- Fishbane, S.N.; Singh, A.K.; Cournoyer, S.H.; Jindal, K.K.; Fanti, P.; Guss, C.D.; Lin, V.H.; Pratt, R.D.; Gupta, A. Ferric pyrophosphate citrate (Triferic™) administration via the dialysate maintains hemoglobin and iron balance in chronic hemodialysis patients. Nephrol. Dial. Transplant. 2015, 30, 2019–2026. [Google Scholar] [CrossRef]
- Macdougall, I.C.; Bock, A.H.; Carrera, F.; Eckardt, K.-U.; Gaillard, C.; Van Wyck, D.; Roubert, B.; Nolen, J.G.; Roger, S.D. On behalf of the FIND-CKD Study Investigators FIND-CKD: A randomized trial of intravenous ferric carboxymaltose versus oral iron in patients with chronic kidney disease and iron deficiency anaemia. Nephrol. Dial. Transplant. 2014, 29, 2075–2084. [Google Scholar] [CrossRef]
- Mursu, J.; Robien, K.; Harnack, L.J.; Park, K.; Jacobs, D.R., Jr. Dietary supplements and mortality rate in older women: The Iowa Women’s Health Study. Arch. Intern. Med. 2011, 171, 1625–1633. [Google Scholar] [CrossRef]
- Rostoker, G.; Griuncelli, M.; Loridon, C.; Magna, T.; Machado, G.; Drahi, G.; Dahan, H.; Janklewicz, P.; Cohen, Y. Reassessment of iron marker for prediction of dialysis iron overload: An MRI study. PLoS ONE 2015, 10, e0132006. [Google Scholar] [CrossRef]
- Anraku, M.; Kitamura, K.; Shintomo, R.; Takeuchi, K.; Ikeda, H.; Nagano, J.; Ko, T.; Mera, K.; Tomita, K.; Otagiri, M. Effect of intravenous iron administration frequency on AOPP and inflammatory biomarkers in chronic hemodialysis patients: A pilot study. Clin. Biochem. 2008, 41, 1168–1174. [Google Scholar] [CrossRef]
- Macdougall, I.C.; White, C.; Anker, S.D.; Bhandari, S.; Farrington, K.; Kalra, P.A.; McMurray, J.J.V.; Murray, H.; Tomson, C.R.V.; Wheeler, D.C.; et al. Intravenous iron in patients undergoing maintenance hemodialysis. N. Engl. J. Med. 2019, 380, 447–458. [Google Scholar] [CrossRef]
- Moradi, Z.; Maali, A.; Shad, J.S.; Farasat, A.; Kouchaki, R.; Moghadami, M.; Ahmadi, M.H.; Azad, M. Updates on Novel Erythropoiesis-Stimulating Agents: Clinical and Molecular Approach. Indian J. Hematol. Blood Transfus. 2020, 36, 26–36. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Hamano, T.; Wada, A.; Masakane, I. Types of Erythropoietin-Stimulating Agents and Mortality among Patients Undergoing Hemodialysis. J. Am. Soc. Nephrol. 2019, 30, 1037–1048. [Google Scholar] [CrossRef]
- Van der Meer, P.; Voors, A.A.; Lipsic, E.; Van Gilst, W.H.; Van Veldhuisen, D.J. Erytropoietin in cardiovascular diseases. Eur. Heart J. 2004, 25, 285–291. [Google Scholar] [CrossRef]
- van der Meer, P.; Lok, D.J.; Januzzi, J.L.; de la Porte, P.W.B.-A.; Lipsic, E.; van Wijngaarden, J.; Voors, A.A.; van Gilst, W.H.; van Veldhuisen, D.J. Adequacy of endogenous erythropoietin levels and mortality in anaemic heart failure patients. Eur. Heart J. 2008, 29, 1510–1515. [Google Scholar] [CrossRef]
- Beverborg, N.G.; van Veldhuisen, D.J.; van der Meer, P. Anemia in Heart Failure: Still Relevant? JACC Heart Fail. 2018, 6, 201–208. [Google Scholar] [CrossRef]
- Locatelli, F.; Barany, P.; Covic, A.; De Francisco, A.; Del Vecchio, L.; Goldsmith, D.; Hörl, W.; London, G.; Vanholder, R.; Van Biesen, W.; et al. Kidney Disease: Improving Global Outcomes guidelines on anaemia management in chronic kidney disease: A European Renal Best Practice position statement. Nephrol. Dial. Transplant. 2013, 28, 1346–1359. [Google Scholar] [CrossRef]
- Hanna, R.M.; Streja, E.; Kalantar-Zadeh, K. Burden of Anemia in Chronic Kidney Disease: Beyond Erythropoietin. Adv. Ther. 2020, 38, 52–75. [Google Scholar] [CrossRef]
- Del Vecchio, L.; Minutolo, R. ESA, Iron Therapy and New Drugs: Are There New Perspectives in the Treatment of Anaemia? J. Clin. Med. 2021, 10, 839. [Google Scholar] [CrossRef]
- Weir, M.R. Managing Anemia across the Stages of Kidney Disease in Those Hyporesponsive to Erythropoiesis-Stimulating Agents. Am. J. Nephrol. 2021, 52, 450–466. [Google Scholar] [CrossRef]
- Silverberg, D.S.; Wexler, D.; Iaina, A. The importance of anemia and its correction in the management of severe congestive heart failure. Eur. J. Heart Fail. 2002, 4, 681–686. [Google Scholar] [CrossRef]
- Ayus, J.C.; Go, A.S.; Valderrabano, F.; Verde, E.; de Vinuesa, S.G.; Achinger, S.G.; Lorenzo, V.; Arieff, A.I.; Luao, J. Spanish Group for the study of the anemia and left ventricular hypertrophy in pre-dialysis patients. Effects of erythropoietin on left ventricular hypertrophy in adults with severe chronic renal failure and hemoglobin < 10 g/dL. Kidney Int. 2005, 68, 788–795. [Google Scholar] [CrossRef]
- Akaishi, M.; Hiroe, M.; Hada, Y.; Suzuki, M.; Tsubakihara, Y.; Akizawa, T. KRN321 Study Group. Effect of anemia correction on left ventricular hypertrophy in patients with modestly high hemoglobin level and chronic kidney disease. J. Cardiol. 2013, 62, 249–256. [Google Scholar] [CrossRef]
- Kuriyam, S.; Tomonari, H.; Yoshida, H.; Hashimoto, T.; Kawaguchi, Y.; Sakai, O. Reversal of Anemia by Erythropoietin Therapy Retards the Progression of Chronic Renal Failure, Especially in Nondiabetic Patients. Nephron 1997, 77, 176–185. [Google Scholar] [CrossRef]
- Gouva, C.; Nikolopoulos, P.; Ioannidis, J.P.; Siamopoulos, K.C. Treating anemia early in renal failure patients slows the decline of renal function: A randomized controlled trial. Kidney Int. 2004, 66, 753–760. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 2013, 3, 1–150. [Google Scholar]
- McCullough, P.A.; Barnhart, H.X.; Inrig, J.K.; Reddan, D.; Sapp, S.; Patel, U.D.; Singh, A.K.; Szczech, L.A.; Califf, R.M. Cardiovascular Toxicity of Epoetin-Alfa in Patients with Chronic Kidney Disease. Am. J. Nephrol. 2013, 37, 549–558. [Google Scholar] [CrossRef]
- Eisenga, M.F.; Emans, M.E.; van der Putten, K.; Cramer, M.J.; Diepenbroek, A.; Velthuis, B.K.; Doevendans, P.A.; Verhaar, M.C.; Joles, J.A.; Bakker, S.J.L.; et al. Epoetin Beta and C-Terminal Fibroblast Growth Factor 23 in Patients with Chronic Heart Failure and Chronic Kidney Disease. J. Am. Heart Assoc. 2019, 8, e011130. [Google Scholar] [CrossRef]
- Jackevicius, C.A.; Co, M.J.; Warner, A.L. Predictors of erythropoietin use in patients with cardiorenal anaemia syndrome. Int. J. Pharm. Pract. 2014, 23, 199–204. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef]
- Haase, V.H. HIF-PH inhibitors for anemia of CKD. Kidney Int. Suppl. 2021, 11, 8–25. [Google Scholar] [CrossRef]
- Fujikawa, R.; Nagao, Y.; Fujioka, M.; Akizawa, T. Treatment of anemia associated with chronic kidney disease with the HIF prolyl hydroxylase inhibitor enarodustat: A review of the evidence. Ther. Apher. Dial. 2022, 26, 679–693. [Google Scholar] [CrossRef]
- Vallon, V.; Rose, M.; Gerasimova, M.; Satriano, J.; Platt, K.A.; Koepsell, H.; Cunard, R.; Sharma, K.; Thomson, S.C.; Rieg, T.; et al. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am. J. Physiol. Physiol. 2013, 304, F156–F167. [Google Scholar] [CrossRef]
- Onishi, A.; Fu, Y.; Patel, R.; Darshi, M.; Crespo-Masip, M.; Huang, W.; Song, P.; Freeman, B.; Kim, Y.C.; Soleimani, M.; et al. A role for the tubular Na+-H+-exchanger NHE3 in the natri-uretic effect of the SGLT2 inhibitor empagliflozin. Am. J. Physiol. Ren. Physiol. 2020, 319, 712–728. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Zinman, B.; Fitchett, D.; Wanner, C.; Ferrannini, E.; Schumacher, M.; Schmoor, C.; Ohneberg, K.; Johansen, O.E.; George, J.T.; et al. How does empagliflozin reduce cardiovascular mortality? Insights from a mediation analysis of the EMPA-REG OUTCOME Trial. Diabetes Care 2018, 41, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Watts, E.R.; Walmsley, S.R. Inflflammation and hypoxia: HIF and PHD isoform selectivity. Trends Mol. Med. 2019, 25, 33–46. [Google Scholar] [CrossRef]
- Iso, T.; Matsue, Y.; Mizukami, A.; Tokano, T.; Isoda, K.; Suwa, S.; Miyauchi, K.; Yanagisawa, N.; Okumura, Y.; Minamino, T. Daprodustat for anaemia in patients with heart failure and chronic kidney disease: A randomized controlled study. ESC Heart Fail. 2022, 9, 4291–4297. [Google Scholar] [CrossRef] [PubMed]
- Sanghani, N.S.; Haase, V.H. Hypoxia-Inducible Factor Activators in Renal Anemia: Current Clinical Experience. Adv. Chronic Kidney Dis. 2019, 26, 253–266. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, Y.; Jia, Y.; Xu, J.; Chai, Y. Roxadustat promotes angiogenesis through HIF-1α/VEGF/VEGFR2 signaling and accelerates cutaneous wound healing in diabetic rats. Wound Repair Regen. 2019, 27, 324–334. [Google Scholar] [CrossRef]
- Chen, N.; Qian, J.; Chen, J.; Yu, X.; Mei, C.; Hao, C.; Jiang, G.; Lin, H.; Zhang, X.; Zuo, L.; et al. Phase 2 studies of oral hypoxia-inducible factor prolyl hydroxylase inhibitor FG-4592 for treatment of anemia in China. Nephrol. Dial. Transplant. 2017, 32, 1373–1386. [Google Scholar] [CrossRef]
- Fishbane, S.; El-Shahawy, M.A.; Pecoits-Filho, R.; Van, B.P.; Houser, M.T.; Frison, L.; Little, D.J.; Guzman, N.J.; Pergola, P.E. Roxadustat for Treating Anemia in Patients with CKD Not on Dialysis: Results from a Randomized Phase 3 Study. J. Am. Soc. Nephrol. 2021, 32, 737–755. [Google Scholar] [CrossRef]
- Provenzano, R.; Shutov, E.; Eremeeva, L.; Korneyeva, S.; Poole, L.; Saha, G.; Bradley, C.; Eyassu, M.; Besarab, A.; Leong, R.; et al. Roxadustat for anemia in patients with end-stage renal disease incident to dialysis. Nephrol. Dial. Transplant. 2021, 36, 1717–1730. [Google Scholar] [CrossRef]
- Chen, N.; Hao, C.; Liu, B.-C.; Lin, H.; Wang, C.; Xing, C.; Liang, X.; Jiang, G.; Liu, Z.; Li, X.; et al. Roxadustat Treatment for Anemia in Patients Undergoing Long-Term Dialysis. N. Engl. J. Med. 2019, 381, 1011–1022. [Google Scholar] [CrossRef]
- Li, Q.Y.; Xiong, Q.W.; Yao, X.; Liu, F.; Tang, X.; Fu, H.; Tong, T.; Mao, J.; Peng, W.X. Roxadustat: Do we know all the answers? Biomol. Biomed. 2023, 23, 354–363. [Google Scholar] [CrossRef] [PubMed]
- PMDA. Japanese Pharmaceutical and Medical Devices Agency Report on Roxadustat. Available online: https://www.pmda.go.jp/files/000234811 (accessed on 22 July 2020).
- Tsubakihara, Y.; Akizawa, T.; Nangaku, M.; Onoue, T.; Yonekawa, T.; Matsushita, H.; Endo, Y.; Cobitz, A. A 24-Week Anemia Correction Study of Daprodustat in Japanese Dialysis Patients. Ther. Apher. Dial. 2019, 24, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Akizawa, T.; MacDougall, I.C.; Berns, J.S.; Bernhardt, T.; Staedtler, G.; Taguchi, M.; Iekushi, K.; Krueger, T. Long-Term Efficacy and Safety of Molidustat for Anemia in Chronic Kidney Disease: DIALOGUE Extension Studies. Am. J. Nephrol. 2019, 49, 271–280. [Google Scholar] [CrossRef]
- Crugliano, G.; Serra, R.; Ielapi, N.; Battaglia, Y.; Coppolino, G.; Bolignano, D.; Bracale, U.M.; Pisani, A.; Faga, T.; Michael, A.; et al. Hypoxia-Inducible Factor Stabilizers in End Stage Kidney Disease: “Can the Promise Be Kept?”. Int. J. Mol. Sci. 2021, 22, 12590. [Google Scholar] [CrossRef]
- Pergola, P.E.; Spinowitz, B.S.; Hartman, C.S.; Maroni, B.J.; Haase, V.H. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis dependent chronic kidney disease. Kidney Int. 2016, 90, 1115–1122. [Google Scholar] [CrossRef]
- Dhillon, S. Desidustat: First Approval. Drugs 2022, 82, 1207–1212. [Google Scholar] [CrossRef]
- Bessho, R.; Takiyama, Y.; Takiyama, T.; Kitsunai, H.; Takeda, Y.; Sakagami, H.; Ota, T. Hypoxia-inducible factor-1α is the therapeutic target of the SGLT2 inhibitor for diabetic nephropathy. Sci. Rep. 2019, 9, 14754. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Eckardt, K.U. HIF prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond. Nat Rev Nephrol. 2016, 12, 157–168. [Google Scholar] [CrossRef]
- Dallalio, G.; Law, E.; Means, R.T. Hepcidin inhibits in vitroerythroid colony formation at reduced erythropoietin concentrations. Blood 2006, 107, 2702–2704. [Google Scholar] [CrossRef]
- Kato, A. Increased hepcidin-25 and erythropoietin responsiveness in patients with cardio-renal anemia syndrome. Future Cardiol. 2010, 6, 769–771. [Google Scholar] [CrossRef]
- Pagani, A.; Nai, A.; Silvestri, L.; Camaschella, C. Hepcidin and Anemia: A Tight Relationship. Front. Physiol. 2019, 10, 1294. [Google Scholar] [CrossRef] [PubMed]
- Srai, S.K.; Chung, B.; Marks, J.; Pourvali, K.; Solanky, N.; Rapisarda, C.; Chaston, T.B.; Hanif, R.; Unwin, R.J.; Debnam, E.S.; et al. Erythropoietin regulates intestinal iron absorption in a rat model of chronic renal failure. Kidney Int. 2010, 78, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Lipsic, E.; Van Der Meer, P.; Van Veldhuisen, D.J. Erythropoiesis-Stimulating Agents and Heart Failure. Cardiovasc. Ther. 2011, 29, e52–e59. [Google Scholar] [CrossRef]
- Park, K.-J.; Jin, H.-M.; Cho, Y.-N.; Kang, J.-H.; Jung, H.-J.; Kang, J.-H.; Kim, J.-E.; Yim, Y.-R.; Lee, J.-W.; Lee, K.-E.; et al. Clinical and Hematological Effects of Tocilizumab on Serum Hepcidin, Anemia Response and Disease Activity in Patients with Active Rheumatoid Arthritis. J. Rheum. Dis. 2016, 23, 37–46. [Google Scholar] [CrossRef]
- Sheetz, M.; Barrington, P.; Callies, S.; Berg, P.; McColm, J.; Marbury, T.; Decker, B.; Dyas, G.L.; Truhlar, S.M.; Benschop, R.; et al. Targeting the hepcidin-ferroportin pathway in anaemia of chronic kidney disease. Br. J. Clin. Pharmacol. 2019, 85, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Renders, L.; Budde, K.; Rosenberger, C.; Van Swelm, R.; Swinkels, D.; Dellanna, F.; Feuerer, W.; Wen, M.; Erley, C.; Bader, B.; et al. First-in-human Phase I studies of PRS-080#22, a hepcidin antagonist, in healthy volunteers and patients with chronic kidney disease undergoing hemodialysis. PLoS ONE 2019, 14, e0212023. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buliga-Finis, O.N.; Ouatu, A.; Tanase, D.M.; Gosav, E.M.; Seritean Isac, P.N.; Richter, P.; Rezus, C. Managing Anemia: Point of Convergence for Heart Failure and Chronic Kidney Disease? Life 2023, 13, 1311. https://doi.org/10.3390/life13061311
Buliga-Finis ON, Ouatu A, Tanase DM, Gosav EM, Seritean Isac PN, Richter P, Rezus C. Managing Anemia: Point of Convergence for Heart Failure and Chronic Kidney Disease? Life. 2023; 13(6):1311. https://doi.org/10.3390/life13061311
Chicago/Turabian StyleBuliga-Finis, Oana Nicoleta, Anca Ouatu, Daniela Maria Tanase, Evelina Maria Gosav, Petronela Nicoleta Seritean Isac, Patricia Richter, and Ciprian Rezus. 2023. "Managing Anemia: Point of Convergence for Heart Failure and Chronic Kidney Disease?" Life 13, no. 6: 1311. https://doi.org/10.3390/life13061311
APA StyleBuliga-Finis, O. N., Ouatu, A., Tanase, D. M., Gosav, E. M., Seritean Isac, P. N., Richter, P., & Rezus, C. (2023). Managing Anemia: Point of Convergence for Heart Failure and Chronic Kidney Disease? Life, 13(6), 1311. https://doi.org/10.3390/life13061311