

Extracellular Vesicles and Cancer Multidrug Resistance: Undesirable Intercellular Messengers?
 ,
,  ,
,
Abstract
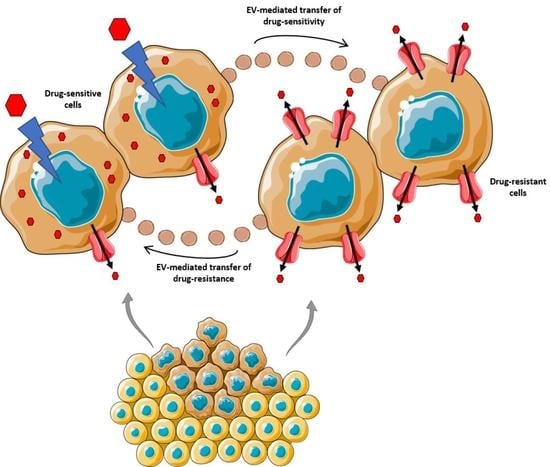
:
1. Introduction
1.1. Multidrug Resistance (MDR)
1.2. ABC Transporters
1.2.1. P-Glycoprotein/ABCB1/MDR1
1.2.2. MRPs/ABCCs
1.2.3. BCRP/ABCG2
1.3. Extracellular Vesicles
2. EVs as Mediators of Multidrug Resistance
2.1. EV-Mediated Shuttle of ABC Transporters
2.2. EV-Mediated Regulation of ABC Transporters
2.2.1. MicroRNAs
Inhibition of Multidrug Resistance via EV-Carried MicroRNAs
Stimulation of Multidrug Resistance via EV-Carried MicroRNAs
2.2.2. Other Noncoding RNAs
2.2.3. Regulatory Proteins
2.3. Drug Sequestration in EVs
3. EVs as Biomarkers of Multidrug Resistance
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The Ever-Increasing Importance of Cancer as a Leading Cause of Premature Death Worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Yu, M.; Xu, J.; Li, B.Y.; Zhao, Y.; Kankala, R.K. Overcoming Cancer Multi-Drug Resistance (MDR): Reasons, Mechanisms, Nanotherapeutic Solutions, and Challenges. Biomed. Pharmacother. 2023, 162, 114643. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Gong, H.; Xu, J.; Huang, Y.; Wu, F.; He, Z. Nanomedicines for Overcoming Cancer Drug Resistance. Pharmaceutics 2022, 14, 1606. [Google Scholar] [CrossRef]
- Ceballos, M.P.; Rigalli, J.P.; Cere, L.I.; Semeniuk, M.; Catania, V.A.; Ruiz, M.L. ABC Transporters: Regulation and Association with Multidrug Resistance in Hepatocellular Carcinoma and Colorectal Carcinoma. Curr. Med. Chem. 2019, 26, 1224–1250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ye, B.; Chen, Z.; Chen, Z.S. Progress in the Studies on the Molecular Mechanisms Associated with Multidrug Resistance in Cancers. Acta Pharm. Sin. B 2023, 13, 982–997. [Google Scholar] [CrossRef]
- Assaraf, Y.G.; Brozovic, A.; Gonçalves, A.C.; Jurkovicova, D.; Linē, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The Multi-Factorial Nature of Clinical Multidrug Resistance in Cancer. Drug Resist. Updates 2019, 46, 100645. [Google Scholar] [CrossRef]
- Yang, L.; Li, A.; Wang, Y.; Zhang, Y. Intratumoral Microbiota: Roles in Cancer Initiation, Development and Therapeutic Efficacy. Signal Transduct. Target. Ther. 2023, 8, 35. [Google Scholar] [CrossRef]
- Alonso-Peña, M.; Sanchez-Martin, A.; Sanchon-Sanchez, P.; Soto-Muñiz, M.; Espinosa-Escudero, R.; Marin, J.J.G. Pharmacogenetics of Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Drug Resist 2019, 2, 680–709. [Google Scholar] [CrossRef]
- Zhou, X.; Ni, Y.; Liang, X.; Lin, Y.; An, B.; He, X.; Zhao, X. Mechanisms of Tumor Resistance to Immune Checkpoint Blockade and Combination Strategies to Overcome Resistance. Front. Immunol. 2022, 13, 915094. [Google Scholar] [CrossRef] [PubMed]
- Panebianco, C.; Andriulli, A.; Pazienza, V. Pharmacomicrobiomics: Exploiting the Drug-Microbiota Interactions in Anticancer Therapies. Microbiome 2018, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Lehouritis, P.; Cummins, J.; Stanton, M.; Murphy, C.T.; McCarthy, F.O.; Reid, G.; Urbaniak, C.; Byrne, W.L.; Tangney, M. Local Bacteria Affect the Efficacy of Chemotherapeutic Drugs. Sci. Rep. 2015, 5, 14554. [Google Scholar] [CrossRef] [Green Version]
- Xuan, C.; Shamonki, J.M.; Chung, A.; DiNome, M.L.; Chung, M.; Sieling, P.A.; Lee, D.J. Microbial Dysbiosis Is Associated with Human Breast Cancer. PLoS ONE 2014, 9, e83744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Su, H.; Johnson, C.H.; Khan, S.A.; Kluger, H.; Lu, L. Intratumour Microbiome Associated with the Infiltration of Cytotoxic CD8+ T Cells and Patient Survival in Cutaneous Melanoma. Eur. J. Cancer 2021, 151, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.J.; Park, A.; Pinkett, H.W. Diversity in ABC Transporters: Type I, II and III Importers. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 426–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckenstaler, R.; Benndorf, R.A. 3D Structure of the Transporter ABCG2—What’s New? Br. J. Pharmacol. 2020, 177, 1485–1496. [Google Scholar] [CrossRef] [Green Version]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC Transporters: The Power to Change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian Drug Efflux Transporters of the ATP Binding Cassette (ABC) Family in Multidrug Resistance: A Review of the Past Decade. Cancer Lett. 2016, 370, 153–164. [Google Scholar] [CrossRef]
- Beek, J.; Guskov, A.; Slotboom, D.J. Structural Diversity of ABC Transporters. J. Gen. Physiol. 2014, 143, 419–435. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.H.; Yu, A.-M. ABC Transporters in Multidrug Resistance and Pharmacokinetics, and Strategies for Drug Development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef]
- De Lange, E.C.M. Multi Drug Resistance P Glycoprotein and Other Transporters. Encycl. Stress 2007, 774–783. [Google Scholar] [CrossRef]
- Glavinas, H.; Krajcsi, P.; Cserepes, J.; Sarkadi, B. The Role of ABC Transporters in Drug Resistance, Metabolism and Toxicity. Curr. Drug Deliv. 2004, 1, 27–42. [Google Scholar] [CrossRef]
- Klaassen, C.D.; Aleksunes, L.M. Xenobiotic, Bile Acid, and Cholesterol Transporters: Function and Regulation. Pharmacol. Rev. 2010, 62, 1–96. [Google Scholar] [CrossRef] [Green Version]
- Juliano, R.L.; Ling, V. A Surface Glycoprotein Modulating Drug Permeability in Chinese Hamster Ovary Cell Mutants. BBA Biomembr. 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Jones, P.M.; George, A.M. The ABC Transporter Structure and Mechanism: Perspectives on Recent Research. Cell. Mol. Life Sci. 2004, 61, 682–699. [Google Scholar] [CrossRef]
- Sharom, F.J. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front. Oncol. 2014, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Katsaros, D.; Wiley, A.; Rigault De La Longrais, I.A.; Puopolo, M.; Yu, H. Expression of MDR1 in Epithelial Ovarian Cancer and Its Association with Disease Progression. Oncol. Res. 2007, 16, 395–403. [Google Scholar] [CrossRef]
- Penson, R.T.; Oliva, E.; Skates, S.J.; Glyptis, T.; Fuller, A.F.; Goodman, A.; Seiden, M.V. Expression of Multidrug Resistance-1 Protein Inversely Correlates with Paclitaxel Response and Survival in Ovarian Cancer Patients: A Study in Serial Samples. Gynecol. Oncol. 2004, 93, 98–106. [Google Scholar] [CrossRef]
- Assaraf, Y.G.; Borgnia, M.J. Differential Reversal of Lipophilic Antifolate Resistance in Mammalian Cells with Modulators of the Multidrug Resistance Phenotype. Anticancer Drugs 1993, 4, 395–406. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug Resistance in Cancer: Role of Atp-Dependent Transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Sarkadi, B.; Homolya, L.; Szakács, G.; Váradi, A. Human Multidrug Resistance ABCB and ABCG Transporters: Participation in a Chemoimmunity Defense System. Physiol. Rev. 2006, 86, 1179–1236. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.F.; Wilhelm, S.; Hibner, B.; Bollag, G. Discovery of a Novel Raf Kinase Inhibitor. Endocr. Relat. Cancer 2001, 8, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Sauna, Z.E.; Smith, M.M.; Müller, M.; Kerr, K.M.; Ambudkar, S.V. The Mechanism of Action of Multidrug-Resistance-Linked P-Glycoprotein. J. Bioenerg. Biomembr. 2001, 33, 481–491. [Google Scholar] [CrossRef]
- Klimecki, W.T.; Futscher, B.W.; Grogan, T.M.; Dalton, W.S. P-Glycoprotein Expression and Function in Circulating Blood Cells from Normal Volunteers. Blood 1994, 83, 2451–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, M.; Paull, K.; Monks, A.; Hose, C.; Lee, J.S.; Weinstein, J.; Grever, M.; Bates, S.; Fojo, T. Generation of a Drug Resistance Profile by Quantitation of Mdr-1/P-Glycoprotein in the Cell Lines of the National Cancer Institute Anticancer Drug Screen. J. Clin. Investig. 1995, 95, 2205–2214. [Google Scholar] [CrossRef]
- Lee, T.D.; Lee, O.W.; Brimacombe, K.R.; Chen, L.; Guha, R.; Lusvarghi, S.; Tebase, B.G.; Klumpp-Thomas, C.; Robey, R.W.; Ambudkar, S.V.; et al. A High-Throughput Screen of a Library of Therapeutics Identifies Cytotoxic Substrates of P-Glycoprotein. Mol. Pharmacol. 2019, 96, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Seelig, A. A General Pattern for Substrate Recognition by P-Glycoprotein. Eur. J. Biochem. 1998, 251, 252–261. [Google Scholar] [CrossRef]
- Yakusheva, E.N.; Titov, D.S. Structure and Function of Multidrug Resistance Protein 1. Biochemistry 2018, 83, 907–929. [Google Scholar] [CrossRef]
- Zhou, S.-F.; Wang, L.-L.; Di, Y.M.; Xue, C.C.; Duan, W.; Li, C.G.; Li, Y. Substrates and Inhibitors of Human Multidrug Resistance Associated Proteins and the Implications in Drug Development. Curr. Med. Chem. 2008, 15, 1981–2039. [Google Scholar] [CrossRef]
- Dinis-Oliveira, R.J.; Remião, F.; Duarte, J.A.; Ferreira, R.; Sánchez Navarro, A.; Bastos, M.L.; Carvalho, F. P-Glycoprotein Induction: An Antidotal Pathway for Paraquat-Induced Lung Toxicity. Free Radic. Biol. Med. 2006, 41, 1213–1224. [Google Scholar] [CrossRef]
- Cascorbi, I. P-Glycoprotein: Tissue Distribution, Substrates, and Functional Consequences of Genetic Variations. Handb. Exp. Pharmacol. 2011, 201, 261–283. [Google Scholar] [CrossRef]
- Choudhuri, S.; Klaassen, C.D. Structure, Function, Expression, Genomic Organization, and Single Nucleotide Polymorphisms of Human ABCB1 (MDR1), ABCC (MRP), and ABCG2 (BCRP) Efflux Transporters. Int. J. Toxicol. 2006, 25, 231–259. [Google Scholar] [CrossRef]
- Baker, E.K.; Johnstone, R.W.; Zalcberg, J.R.; El-Osta, A. Epigenetic Changes to the MDR1 Locus in Response to Chemotherapeutic Drugs. Oncogene 2005, 24, 8061–8075. [Google Scholar] [CrossRef] [Green Version]
- Scotto, K.W. Transcriptional Regulation of ABC Drug Transporters. Oncogene 2003, 22, 7496–7511. [Google Scholar] [CrossRef] [Green Version]
- Burk, O.; Arnold, K.A.; Geick, A.; Tegude, H.; Eichelbaum, M. A Role for Constitutive Androstane Receptor in the Regulation of Human Intestinal MDR1 Expression. Biol. Chem. 2005, 386, 503–513. [Google Scholar] [CrossRef]
- Rigalli, J.P.; Ciriaci, N.; Arias, A.; Ceballos, M.P.; Villanueva, S.S.M.; Luquita, M.G.; Mottino, A.D.; Ghanem, C.I.I.; Catania, V.A.; Ruiz, M.L. Regulation of Multidrug Resistance Proteins by Genistein in a Hepatocarcinoma Cell Line: Impact on Sorafenib Cytotoxicity. PLoS ONE 2015, 10, e0119502. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.F.; Qiu, W.; Liu, Z.Q.; Zhu, L.J.; Liu, Z.Q.; Tu, J.H.; Wang, D.; Li, Z.; He, J.; Zhong, G.P.; et al. Effects of Genetic Polymorphisms of CYP3A4, CYP3A5 and MDR1 on Cyclosporine Pharmacokinetics after Renal Transplantation. Clin. Exp. Pharmacol. Physiol. 2006, 33, 1093–1098. [Google Scholar] [CrossRef]
- Jing, W.; Safarpour, Y.; Zhang, T.; Guo, P.; Chen, G.; Wu, X.; Fu, Q.; Wang, Y. Berberine Upregulates P-Glycoprotein in Human Caco-2 Cells and in an Experimental Model of Colitis in the Rat via Activation of Nrf2-Dependent Mechanismss. J. Pharmacol. Exp. Ther. 2018, 366, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Brayboy, L.M.; Knapik, L.O.; Long, S.; Westrick, M.; Wessel, G.M. Ovarian Hormones Modulate Multidrug Resistance Transporters in the Ovary. Contracept. Reprod. Med. 2018, 3, 26. [Google Scholar] [CrossRef] [Green Version]
- Mutoh, K.; Tsukahara, S.; Mitsuhashi, J.; Katayama, K.; Sugimoto, Y. Estrogen-Mediated Post Transcriptional down-Regulation of P-Glycoprotein in MDR1-Transduced Human Breast Cancer Cells. Cancer Sci. 2006, 97, 1198–1204. [Google Scholar] [CrossRef]
- Toscano-Garibay, J.D.; Aquino-Jarquin, G. Regulation Exerted by MiRNAs in the Promoter and UTR Sequences: MDR1/P-Gp Expression as a Particular Case. DNA Cell Biol. 2012, 31, 1358–1364. [Google Scholar] [CrossRef] [PubMed]
- Yague, E.; Armesilla, A.L.; Harrison, G.; Elliott, J.; Sardini, A.; Higgins, C.F.; Raguz, S. P-Glycoprotein (MDR1) Expression in Leukemic Cells Is Regulated at Two Distinct Steps, MRNA Stabilization and Translational Initiation. J. Biol. Chem. 2003, 278, 10344–10352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ménez, C.; Mselli-Lakhal, L.; Foucaud-Vignault, M.; Balaguer, P.; Alvinerie, M.; Lespine, A. Ivermectin Induces P-Glycoprotein Expression and Function through MRNA Stabilization in Murine Hepatocyte Cell Line. Biochem. Pharmacol. 2012, 83, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; De Haa, M.; Scheffer, G.L.; Scheper, R.J.; Van Eijk, M.J.T.; Juijn, J.A. Analysis of Expression of CMOAT (MRP2), MRP3, MRP4, and MRP5, Homologues of the Multidrug Resistance-Associated Protein Gene (MRP1), in Human Cancer Cell Lines. Cancer Res. 1997, 57, 3537–3547. [Google Scholar]
- Hopper, E.; Belinsky, M.G.; Zeng, H.; Tosolini, A.; Testa, J.R.; Kruh, G.D. Analysis of the Structure and Expression Pattern of MRP7 (ABCC10), a New Member of the MRP Subfamily. Cancer Lett. 2001, 162, 181–191. [Google Scholar] [CrossRef]
- Bera, T.K.; Lee, S.; Salvatore, G.; Lee, B.; Pastan, I. MRP8, A New Member of ABC Transporter Superfamily, Identified by EST Database Mining and Gene Prediction Program, Is Highly Expressed in Breast Cancer. Mol. Med. 2001, 7, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Hirohashi, T.; Suzuki, H.; Sugiyama, Y. Characterization of the Transport Properties of Cloned Rat Multidrug Resistance-Associated Protein 3 (MRP3). J. Biol. Chem. 1999, 274, 15181–15185. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.P.; Bhardwaj, G.; Gerlach, J.H.; Mackie, J.E.; Grant, C.E.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.; Deeley, R.G. Overexpression of a Transporter Gene in a Multidrug-Resistant Human Lung Cancer Cell Line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef]
- Kunická, T.; Souček, P. Importance of ABCC1 for Cancer Therapy and Prognosis. Drug Metab. Rev. 2014, 46, 325–342. [Google Scholar] [CrossRef]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. A Family of Drug Transporters: The Multidrug Resistance-Associated Proteins. J. Natl. Cancer Inst. 2000, 92, 1295–1302. [Google Scholar] [CrossRef]
- Büchler, M.; König, J.; Brom, M.; Kartenbeck, J.; Spring, H.; Horie, T.; Keppler, D. CDNA Cloning of the Hepatocyte Canalicular Isoform of the Multidrug Resistance Protein, CMrp, Reveals a Novel Conjugate Export Pump Deficient in Hyperbilirubinemic Mutant Rats. J. Biol. Chem. 1996, 271, 15091–15098. [Google Scholar] [CrossRef] [Green Version]
- König, J.; Rost, D.; Cui, Y.; Keppler, D. Characterization of the Human Multidrug Resistance Protein Isoform MRP3 Localized to the Basolateral Hepatocyte Membrane. Hepatology 1999, 29, 1156–1163. [Google Scholar] [CrossRef]
- Zelcer, N.; van de Wetering, K.; Hillebrand, M.; Sarton, E.; Kuil, A.; Wielinga, P.R.; Tephly, T.; Dahan, A.; Beijnen, J.H.; Borst, P. Mice Lacking Multidrug Resistance Protein 3 Show Altered Morphine Pharmacokinetics and Morphine-6-Glucuronide Antinociception. Proc. Natl. Acad. Sci. USA 2005, 102, 7274–7279. [Google Scholar] [CrossRef]
- Kuroda, M.; Kobayashi, Y.; Tanaka, Y.; Itani, T.; Mifuji, R.; Araki, J.; Kaito, M.; Adachi, Y. Increased Hepatic and Renal Expressions of Multidrug Resistance-Associated Protein 3 in Eisai Hyperbilirubinuria Rats. J. Gastroenterol. Hepatol. 2004, 19, 146–153. [Google Scholar] [CrossRef]
- Rost, D.; König, J.; Weiss, G.; Klar, E.; Stremmel, W.; Keppler, D. Expression and Localization of the Multidrug Resistance Proteins MRP2 and MRP3 in Human Gallbladder Epithelia. Gastroenterology 2001, 121, 1203–1208. [Google Scholar] [CrossRef]
- Borst, P.; Zelcer, N.; van de Wetering, K. MRP2 and 3 in Health and Disease. Cancer Lett. 2006, 234, 51–61. [Google Scholar] [CrossRef]
- Zollner, G.; Wagner, M.; Fickert, P.; Silbert, D.; Fuchsbichler, A.; Zatloukal, K.; Denk, H.; Trauner, M. Hepatobiliary Transporter Expression in Human Hepatocellular Carcinoma. Liver Int. 2005, 25, 367–379. [Google Scholar] [CrossRef]
- Benderra, Z.; Faussat, A.M.; Sayada, L.; Perrot, J.-Y.; Tang, R.; Chaoui, D.; Morjani, H.; Marzac, C.; Marie, J.-P.; Legrand, O. MRP3, BCRP, and P-Glycoprotein Activities Are Prognostic Factors in Adult Acute Myeloid Leukemia. Clin. Cancer Res. 2005, 11, 7764–7772. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Ohnuma, S.; V. Ambudkar, S. Improving Cancer Chemotherapy with Modulators of ABC Drug Transporters. Curr. Drug Targets 2011, 12, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Shibayama, Y.; Nakano, K.; Maeda, H.; Taguchi, M.; Ikeda, R.; Sugawara, M.; Iseki, K.; Takeda, Y.; Yamada, K. Multidrug Resistance Protein 2 Implicates Anticancer Drug-Resistance to Sorafenib. Biol. Pharm. Bull. 2011, 34, 433–435. [Google Scholar] [CrossRef] [Green Version]
- Tomonari, T.; Takeishi, S.; Taniguchi, T.; Tanaka, T. MRP3 as a Novel Resistance Factor for Sorafenib in Hepatocellular Carcinoma. Oncotarget 2016, 7, 7207–7215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuetz, J.D.; Connelly, M.C.; Sun, D.; Paibir, S.G.; Flynn, P.M.; Srinivas, R.V.; Kumar, A.; Fridland, A. MRP4: A Previously Unidentified Factor in Resistance to Nucleoside-Based Antiviral Drugs. Nat. Med. 1999, 5, 1048–1051. [Google Scholar] [CrossRef] [PubMed]
- Wijnholds, J.; Mol, C.A.; van Deemter, L.; de Haas, M.; Scheffer, G.L.; Baas, F.; Beijnen, J.H.; Scheper, R.J.; Hatse, S.; De Clercq, E.; et al. Multidrug-Resistance Protein 5 Is a Multispecific Organic Anion Transporter Able to Transport Nucleotide Analogs. Proc. Natl. Acad. Sci. USA 2000, 97, 7476–7481. [Google Scholar] [CrossRef] [PubMed]
- Lee, K. Analysis of the MRP4 Drug Resistance Profile in Transfected NIH3T3 Cells. J. Natl. Cancer Inst. 2000, 92, 1934–1940. [Google Scholar] [CrossRef]
- Kurz, E.U.; Cole, S.P.; Deeley, R.G. Identification of DNA-Protein Interactions in the 5′ Flanking and 5′ Untranslated Regions of the Human Multidrug Resistance Protein (MRP1) Gene: Evaluation of a Putative Antioxidant Response Element/AP-1 Binding Site. Biochem. Biophys. Res. Commun. 2001, 285, 981–990. [Google Scholar] [CrossRef]
- Manohar, C.F.; Bray, J.A.; Salwen, H.R.; Madafiglio, J.; Cheng, A.; Flemming, C.; Marshall, G.M.; Norris, M.D.; Haber, M.; Cohn, S.L. MYCN-Mediated Regulation of the MRP1 Promoter in Human Neuroblastoma. Oncogene 2004, 23, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Si, X.; Gao, Z.; Xu, F.; Zheng, Y. SOX2 Upregulates Side Population Cells and Enhances Their Chemoresistant Ability by Transactivating ABCC1 Expression Contributing to Intrinsic Resistance to Paclitaxel in Melanoma. Mol. Carcinog. 2020, 59, 257–264. [Google Scholar] [CrossRef]
- Wei, L.; Lin, Q.; Lu, Y.; Li, G.; Huang, L.; Fu, Z.; Chen, R.; Zhou, Q. Cancer-Associated Fibroblasts-Mediated ATF4 Expression Promotes Malignancy and Gemcitabine Resistance in Pancreatic Cancer via the TGF-Β1/SMAD2/3 Pathway and ABCC1 Transactivation. Cell Death Dis. 2021, 12, 334. [Google Scholar] [CrossRef]
- Ji, L.; Li, H.; Gao, P.; Shang, G.; Zhang, D.D.; Zhang, N.; Jiang, T. Nrf2 Pathway Regulates Multidrug-Resistance-Associated Protein 1 in Small Cell Lung Cancer. PLoS ONE 2013, 8, e63404. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, M.L.; Villanueva, S.S.M.; Luquita, M.G.; Pellegrino, J.M.; Rigalli, J.P.; Arias, A.; Sánchez Pozzi, E.J.; Mottino, A.D.; Catania, V.A. Induction of Intestinal Multidrug Resistance-Associated Protein 2 (Mrp2) by Spironolactone in Rats. Eur. J. Pharmacol. 2009, 623, 103–106. [Google Scholar] [CrossRef]
- di Masi, A.; De Marinis, E.; Ascenzi, P.; Marino, M. Nuclear Receptors CAR and PXR: Molecular, Functional, and Biomedical Aspects. Mol. Asp. Med. 2009, 30, 297–343. [Google Scholar] [CrossRef]
- Johnson, D.R.; Klaassen, C.D. Regulation of Rat Multidrug Resistance Protein 2 by Classes of Prototypical Microsomal Enzyme Inducers that Activate Distinct Transcription Pathways. Toxicol. Sci. 2002, 67, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Kast, H.R.; Goodwin, B.; Tarr, P.T.; Jones, S.A.; Anisfeld, A.M.; Stoltz, C.M.; Tontonoz, P.; Kliewer, S.; Willson, T.M.; Edwards, P.A. Regulation of Multidrug Resistance-Associated Protein 2 (ABCC2) by the Nuclear Receptors Pregnane X Receptor, Farnesoid X-Activated Receptor, and Constitutive Androstane Receptor. J. Biol. Chem. 2002, 277, 2908–2915. [Google Scholar] [CrossRef] [Green Version]
- Stöckel, B.; König, J.; Nies, A.T.; Cui, Y.; Brom, M.; Keppler, D. Characterization of the 5′-Flanking Region of the Human Multidrug Resistance Protein 2 (MRP2) Gene and Its Regulation in Comparison with the Multidrug Resistance Protein 3 (MRP3) Gene. Eur. J. Biochem. 2000, 267, 1347–1358. [Google Scholar] [CrossRef] [Green Version]
- Vollrath, V.; Wielandt, A.M.; Iruretagoyena, M.; Chianale, J. Role of Nrf2 in the Regulation of the Mrp2 (ABCC2) Gene. Biochem. J. 2006, 395, 599–609. [Google Scholar] [CrossRef] [Green Version]
- Rigalli, J.P.; Perdomo, V.G.; Ciriaci, N.; Francés, D.E.A.; Ronco, M.T.; Bataille, A.M.; Ghanem, C.I.; Ruiz, M.L.; Manautou, J.E.; Catania, V.A. The Trypanocidal Benznidazole Promotes Adaptive Response to Oxidative Injury: Involvement of the Nuclear Factor-Erythroid 2-Related Factor-2 (Nrf2) and Multidrug Resistance Associated Protein 2 (MRP2). Toxicol. Appl. Pharmacol. 2016, 304, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Hirai, T.; Fukui, Y.; Motojima, K. PPARalpha Agonists Positively and Negatively Regulate the Expression of Several Nutrient/Drug Transporters in Mouse Small Intestine. Biol. Pharm. Bull. 2007, 30, 2185–2190. [Google Scholar] [CrossRef] [Green Version]
- Trauner, M. Molecular Alterations of Canalicular Transport Systems in Experimental Models of Cholestasis: Possible Functional Correlations. Yale J. Biol. Med. 1997, 70, 365–378. [Google Scholar]
- Cao, J.; Huang, L.; Liu, Y.; Hoffman, T.; Stieger, B.; Meier, P.J.; Vore, M. Differential Regulation of Hepatic Bile Salt and Organic Anion Transporters in Pregnant and Postpartum Rats and the Role of Prolactin. Hepatology 2001, 33, 140–147. [Google Scholar] [CrossRef]
- Haenisch, S.; Laechelt, S.; Bruckmueller, H.; Werk, A.; Noack, A.; Bruhn, O.; Remmler, C.; Cascorbi, I. Down-Regulation of ATP-Binding Cassette C2 Protein Expression in HepG2 Cells after Rifampicin Treatment Is Mediated by MicroRNA-379. Mol. Pharmacol. 2011, 80, 314–320. [Google Scholar] [CrossRef]
- Tian, J.; Xu, Y.-Y.; Li, L.; Hao, Q. MiR-490-3p Sensitizes Ovarian Cancer Cells to Cisplatin by Directly Targeting ABCC2. Am. J. Transl. Res. 2017, 9, 1127–1138. [Google Scholar] [PubMed]
- Molina-Pinelo, S.; Gutiérrez, G.; Pastor, M.D.; Hergueta, M.; Moreno-Bueno, G.; García-Carbonero, R.; Nogal, A.; Suárez, R.; Salinas, A.; Pozo-Rodríguez, F.; et al. MicroRNA-Dependent Regulation of Transcription in Non-Small Cell Lung Cancer. PLoS ONE 2014, 9, e90524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A Multidrug Resistance Transporter from Human MCF-7 Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef]
- Liu, F.-S. Mechanisms of Chemotherapeutic Drug Resistance in Cancer Therapy—A Quick Review. Taiwan J. Obstet. Gynecol. 2009, 48, 239–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.G.; Gump, J.L.; Zhang, C.; Cook, J.M.; Marchion, D.; Hazlehurst, L.; Munster, P.; Schell, M.J.; Dalton, W.S.; Sullivan, D.M. ABCG2 Expression, Function, and Promoter Methylation in Human Multiple Myeloma. Blood 2006, 108, 3881–3889. [Google Scholar] [CrossRef]
- Glavinas, H.; Krajcsi, P.; Cserepes, J.; Sarkadi, B. The Role of ABC Transporters in Drug Absorption, Distribution, Metabolism, Excretion and Toxicity (ADME-Tox). Curr. Drug Deliv. 2008, 1, 379–393. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug Resistance in Cancer: An Overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Kim, B.; Fatayer, H.; Hanby, A.M.; Horgan, K.; Perry, S.L.; Valleley, E.M.A.; Verghese, E.T.; Williams, B.J.; Thorne, J.L.; Hughes, T.A. Neoadjuvant Chemotherapy Induces Expression Levels of Breast Cancer Resistance Protein that Predict Disease-Free Survival in Breast Cancer. PLoS ONE 2013, 8, e62766. [Google Scholar] [CrossRef] [Green Version]
- Calcagno, A.M.; Fostel, J.M.; To, K.K.W.; Salcido, C.D.; Martin, S.E.; Chewning, K.J.; Wu, C.-P.; Varticovski, L.; Bates, S.E.; Caplen, N.J.; et al. Single-Step Doxorubicin-Selected Cancer Cells Overexpress the ABCG2 Drug Transporter through Epigenetic Changes. Br. J. Cancer 2008, 98, 1515–1524. [Google Scholar] [CrossRef] [Green Version]
- To, K.K.W.; Zhan, Z.; Bates, S.E. Aberrant Promoter Methylation of the ABCG2 Gene in Renal Carcinoma. Mol. Cell. Biol. 2006, 26, 8572–8585. [Google Scholar] [CrossRef] [Green Version]
- To, K.K.W.; Zhan, Z.; Litman, T.; Bates, S.E. Regulation of ABCG2 Expression at the 3′ Untranslated Region of Its MRNA through Modulation of Transcript Stability and Protein Translation by a Putative MicroRNA in the S1 Colon Cancer Cell Line. Mol. Cell. Biol. 2008, 28, 5147–5161. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.-T.; He, M.; Wang, Y.; Jiao, X.-Y.; Zhao, L.; Bai, X.-F.; Yu, Z.-J.; Wu, H.-Z.; Sun, M.-L.; Song, Z.-G.; et al. MiR-487a Resensitizes Mitoxantrone (MX)-Resistant Breast Cancer Cells (MCF-7/MX) to MX by Targeting Breast Cancer Resistance Protein (BCRP/ABCG2). Cancer Lett. 2013, 339, 107–115. [Google Scholar] [CrossRef]
- D’Souza-Schorey, C.; Schorey, J.S. Regulation and Mechanisms of Extracellular Vesicle Biogenesis and Secretion. Essays Biochem. 2018, 62, 125–133. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Yates, A.G.; Pink, R.C.; Erdbrügger, U.; Siljander, P.R.-M.; Dellar, E.R.; Pantazi, P.; Akbar, N.; Cooke, W.R.; Vatish, M.; Dias-Neto, E.; et al. In Sickness and in Health: The Functional Role of Extracellular Vesicles in Physiology and Pathology in Vivo. Part I: Health and Normal Physiology. J. Extracell. Vesicles 2022, 11, e12151. [Google Scholar] [CrossRef]
- Yates, A.G.; Pink, R.C.; Erdbrügger, U.; Siljander, P.R.-M.; Dellar, E.R.; Pantazi, P.; Akbar, N.; Cooke, W.R.; Vatish, M.; Dias-Neto, E.; et al. In Sickness and in Health: The Functional Role of Extracellular Vesicles in Physiology and Pathology in Vivo. Part II: Pathology. J. Extracell. Vesicles 2022, 11, e12190. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.-A.; Gu, N.-Y.; Jeong, S.Y.; Byeon, J.S.; Jeong, D.-U.; Ouh, I.-O.; Lee, Y.-H.; Hyun, B.-H. Canine Natural Killer Cell-Derived Exosomes Exhibit Antitumor Activity in a Mouse Model of Canine Mammary Tumor. Biomed Res. Int. 2021, 2021, 6690704. [Google Scholar] [CrossRef]
- Sousa, D.; Lima, R.T.; Lopes-Rodrigues, V.; Gonzalez, E.; Royo, F.; Xavier, C.P.R.; Falcón-Pérez, J.M.; Vasconcelos, M.H. Different Ability of Multidrug-Resistant and -Sensitive Counterpart Cells to Release and Capture Extracellular Vesicles. Cells 2021, 10, 2886. [Google Scholar] [CrossRef]
- Lopes-Rodrigues, V.; Di Luca, A.; Sousa, D.; Seca, H.; Meleady, P.; Henry, M.; Lima, R.T.; O’Connor, R.; Vasconcelos, M.H. Multidrug Resistant Tumour Cells Shed More Microvesicle-like EVs and Less Exosomes than Their Drug-Sensitive Counterpart Cells. Biochim. Biophys. Acta 2016, 1860, 618–627. [Google Scholar] [CrossRef]
- Berguetti, T.S.; Quintaes, L.S.P.; Hancio Pereira, T.; Robaina, M.; Cruz, A.; Maia, R.C.; de Souza, P. TNF-α Modulates P-Glycoprotein Expression and Contributes to Cellular Proliferation via Extracellular Vesicles. Cells 2019, 8, 500. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, J.; Galas, L.; Boulangé-Lecomte, C.; Rioult, D.; Bultelle, F.; Magal, P.; Webb, G.; Le Foll, F. Different Modalities of Intercellular Membrane Exchanges Mediate Cell-to-Cell p-Glycoprotein Transfers in MCF-7 Breast Cancer Cells. J. Biol. Chem. 2012, 287, 7374–7387. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xu, C.; Hua, Y.; Sun, L.; Cheng, K.; Jia, Z.; Han, Y.; Dong, J.; Cui, Y.; Yang, Z. Exosomes Play an Important Role in the Process of Psoralen Reverse Multidrug Resistance of Breast Cancer. J. Exp. Clin. Cancer Res. 2016, 35, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, M.; Zhu, X.; Chen, W.; Zhong, S.; Hu, Q.; Ma, T.; Zhang, J.; Chen, L.; Tang, J.; Zhao, J. Exosomes Mediate Drug Resistance Transfer in MCF-7 Breast Cancer Cells and a Probable Mechanism Is Delivery of P-Glycoprotein. Tumor Biol. 2014, 35, 10773–10779. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.; Gong, J.; Sambasivam, S.; Combes, V.; Mathys, J.-M.; Davey, R.; Grau, G.E.R.; Bebawy, M. Microparticle-associated Nucleic Acids Mediate Trait Dominance in Cancer. FASEB J. 2012, 26, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.; Luk, F.; Dalla, P.V.; Grau, G.E.R.; Bebawy, M. Breast Cancer-Derived Microparticles Display Tissue Selectivity in the Transfer of Resistance Proteins to Cells. PLoS ONE 2013, 8, e61515. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Miao, K.; Sun, H.; Deng, C.-X. Tumor Heterogeneity Reshapes the Tumor Microenvironment to Influence Drug Resistance. Int. J. Biol. Sci. 2022, 18, 3019–3033. [Google Scholar] [CrossRef]
- Bebawy, M.; Combes, V.; Lee, E.; Jaiswal, R.; Gong, J.; Bonhoure, A.; Grau, G.E.R. Membrane Microparticles Mediate Transfer of P-Glycoprotein to Drug Sensitive Cancer Cells. Leukemia 2009, 23, 1643–1649. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Qiao, D.; Chen, L.; Xu, M.; Chen, S.; Huang, L.; Wang, F.; Chen, Z.; Cai, J.; Fu, L. Chemotherapeutic Drugs Stimulate the Release and Recycling of Extracellular Vesicles to Assist Cancer Cells in Developing an Urgent Chemoresistance. Mol. Cancer 2019, 18, 182. [Google Scholar] [CrossRef] [Green Version]
- Cai, T.; Zhang, C.; Zhan, T. Transfer of Exosomal MicroRNAs Confers Doxorubicin Resistance in Osteosarcoma Cells. Mol. Med. Rep. 2023, 27, 86. [Google Scholar] [CrossRef]
- Torreggiani, E.; Roncuzzi, L.; Perut, F.; Zini, N.; Baldini, N. Multimodal Transfer of MDR by Exosomes in Human Osteosarcoma. Int. J. Oncol. 2016, 49, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Schirizzi, A.; Contino, M.; Carrieri, L.; Riganti, C.; De Leonardis, G.; Scavo, M.P.; Perrone, M.G.; Miciaccia, M.; Kopecka, J.; Refolo, M.G.; et al. The Multiple Combination of Paclitaxel, Ramucirumab and Elacridar Reverses the Paclitaxel-Mediated Resistance in Gastric Cancer Cell Lines. Front. Oncol. 2023, 13, 1129832. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; He, J.; Zou, J.; Yu, S.; Sun, X.; Qin, L. Cisplatin-Resistant HepG2 Cell-Derived Exosomes Transfer Cisplatin Resistance to Cisplatin-Sensitive Cells in HCC. PeerJ 2021, 9, e11200. [Google Scholar] [CrossRef] [PubMed]
- Osteikoetxea, X.; Benke, M.; Rodriguez, M.; Pálóczi, K.; Sódar, B.W.; Szvicsek, Z.; Szabó-Taylor, K.; Vukman, K.V.; Kittel, Á.; Wiener, Z.; et al. Detection and Proteomic Characterization of Extracellular Vesicles in Human Pancreatic Juice. Biochem. Biophys. Res. Commun. 2018, 499, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, C.; Rani, S.; O’Brien, K.; O’Neill, A.; Prencipe, M.; Sheikh, R.; Webb, G.; McDermott, R.; Watson, W.; Crown, J.; et al. Docetaxel-Resistance in Prostate Cancer: Evaluating Associated Phenotypic Changes and Potential for Resistance Transfer via Exosomes. PLoS ONE 2012, 7, e50999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.-S.; Choi, D.-Y.; Hong, B.S.; Jang, S.C.; Kim, D.-K.; Lee, J.; Kim, Y.-K.; Kim, K.P.; Gho, Y.S. Quantitative Proteomics of Extracellular Vesicles Derived from Human Primary and Metastatic Colorectal Cancer Cells. J. Extracell. Vesicles 2012, 1, 18704. [Google Scholar] [CrossRef]
- Cao, D.; Qin, S.; Mu, Y.; Zhong, M. The Role of MRP1 in the Multidrug Resistance of Colorectal Cancer. Oncol. Lett. 2017, 13, 2471–2476. [Google Scholar] [CrossRef] [Green Version]
- Kryczka, J.; Sochacka, E.; Papiewska-Pająk, I.; Boncela, J. Implications of ABCC4-Mediated CAMP Eflux for CRC Migration. Cancers 2020, 12, 3547. [Google Scholar] [CrossRef]
- Lu, J.F.; Luk, F.; Gong, J.; Jaiswal, R.; Grau, G.E.R.; Bebawy, M. Microparticles Mediate MRP1 Intercellular Transfer and the Re-Templating of Intrinsic Resistance Pathways. Pharmacol. Res. 2013, 76, 77–83. [Google Scholar] [CrossRef]
- Bouvy, C.; Wannez, A.; Laloy, J.; Chatelain, C.; Dogné, J.-M. Transfer of Multidrug Resistance among Acute Myeloid Leukemia Cells via Extracellular Vesicles and Their MicroRNA Cargo. Leuk. Res. 2017, 62, 70–76. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, L.; Zhu, Y.; Chen, Z.; Qi, X.; Jin, L.; Jin, J.; Hua, D.; Ma, X. Breast Cancer Resistance Protein (BCRP)-Containing Circulating Microvesicles Contribute to Chemoresistance in Breast Cancer. Oncol. Lett. 2015, 10, 3742–3748. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.N.; He, Q.; Wang, G.; Dasgupta, S.; Dinkins, M.B.; Zhu, G.; Kim, A.; Spassieva, S.; Bieberich, E. Guggulsterone and Bexarotene Induce Secretion of Exosome-Associated Breast Cancer Resistance Protein and Reduce Doxorubicin Resistance in MDA-MB-231 Cells. Int. J. Cancer 2015, 137, 1610–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barzegar, M.; Allahbakhshian Farsan, M.; Amiri, V.; Mohammadi, S.; Shahsavan, S.; Mirzaeian, A.; Mohammadi, M.H. AML-Derived Extracellular Vesicles Confer De Novo Chemoresistance to Leukemic Myeloblast Cells by Promoting Drug Export Genes Expression and ROS Inhibition. Iran. J. Pharm. Res. 2021, 20, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.F.; Pokharel, D.; Bebawy, M. A Novel Mechanism Governing the Transcriptional Regulation of ABC Transporters in MDR Cancer Cells. Drug Deliv. Transl. Res. 2017, 7, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhang, Y.; Guo, L.; Liu, C.; Wang, P.; Ren, W. Exosomal MicroRNA-107 Reverses Chemotherapeutic Drug Resistance of Gastric Cancer Cells through HMGA2/MTOR/P-Gp Pathway. BMC Cancer 2021, 21, 1290. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Zhu, W.; Kan, X.; Li, L.; Wu, D. Luteolin Attenuates the Chemoresistance of Osteosarcoma through Inhibiting the PTN/β-Catenin/MDR1 Signaling Axis by Upregulating MiR-384. J. Bone Oncol. 2022, 34, 100429. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, H.; Yao, Y.-F.; Zhong, S.-L.; Zhao, J.H.; Tang, J.H. β-Elemene Reverses Chemoresistance of Breast Cancer Cells by Reducing Resistance Transmission via Exosomes. Cell. Physiol. Biochem. 2015, 36, 2274–2286. [Google Scholar] [CrossRef] [Green Version]
- Semaan, L.; Zeng, Q.; Lu, Y.; Zhang, Y.; Zreik, M.M.; Chamseddine, M.B.; Chopp, M.; Zhang, Z.G.; Moonka, D. MicroRNA-214 Enriched Exosomes from Human Cerebral Endothelial Cells (HCEC) Sensitize Hepatocellular Carcinoma to Anti-Cancer Drugs. Oncotarget 2021, 12, 185–198. [Google Scholar] [CrossRef]
- Munoz, J.L.; Bliss, S.A.; Greco, S.J.; Ramkissoon, S.H.; Ligon, K.L.; Rameshwar, P. Delivery of Functional Anti-MiR-9 by Mesenchymal Stem Cell-Derived Exosomes to Glioblastoma Multiforme Cells Conferred Chemosensitivity. Mol. Ther. Nucleic Acids 2013, 2, e126. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, Y.; Ye, M.; Wu, J.; Ma, L.; Chen, H. Cisplatin-Resistant MDA-MB-231 Cell-Derived Exosomes Increase the Resistance of Recipient Cells in an Exosomal MiR-423-5p-Dependent Manner. Curr. Drug Metab. 2019, 20, 804–814. [Google Scholar] [CrossRef]
- Shen, M.; Dong, C.; Ruan, X.; Yan, W.; Cao, M.; Pizzo, D.; Wu, X.; Yang, L.; Liu, L.; Ren, X.; et al. Chemotherapy-Induced Extracellular Vesicle MiRNAs Promote Breast Cancer Stemness by Targeting ONECUT2. Cancer Res. 2019, 79, 3608–3621. [Google Scholar] [CrossRef]
- Lucotti, S.; Rainaldi, G.; Evangelista, M.; Rizzo, M. Fludarabine Treatment Favors the Retention of MiR-485-3p by Prostate Cancer Cells: Implications for Survival. Mol. Cancer 2013, 12, 52. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Lin, L.; Liu, Q.; Gao, W.; Chen, L.; Sha, C.; Chen, Q.; Xu, W.; Li, Y.; Zhu, X. Exosomal Transfer of MiR-429 Confers Chemoresistance in Epithelial Ovarian Cancer. Am. J. Cancer Res. 2021, 11, 2124–2141. [Google Scholar]
- Fu, X.; Liu, M.; Qu, S.; Ma, J.; Zhang, Y.; Shi, T.; Wen, H.; Yang, Y.; Wang, S.; Wang, J.; et al. Exosomal MicroRNA-32-5p Induces Multidrug Resistance in Hepatocellular Carcinoma via the PI3K/Akt Pathway. J. Exp. Clin. Cancer Res. 2018, 37, 52. [Google Scholar] [CrossRef]
- Zhu, T.; Hu, Z.; Wang, Z.; Ding, H.; Li, R.; Wang, J.; Wang, G. MicroRNA-301b-3p from Mesenchymal Stem Cells-Derived Extracellular Vesicles Inhibits TXNIP to Promote Multidrug Resistance of Gastric Cancer Cells. Cell Biol. Toxicol. 2022. [Google Scholar] [CrossRef]
- Gao, X.; Zhou, J.; Wang, J.; Dong, X.; Chang, Y.; Jin, Y. Mechanism of Exosomal MiR-155 Derived from Bone Marrow Mesenchymal Stem Cells on Stemness Maintenance and Drug Resistance in Myeloma Cells. J. Orthop. Surg. Res. 2021, 16, 637. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, J.; Liu, N.; Wu, W.; Li, H.; Chen, J.; Guo, X. Molecular Mechanism of CD163(+) Tumor-Associated Macrophage (TAM)-Derived Exosome-Induced Cisplatin Resistance in Ovarian Cancer Ascites. Ann. Transl. Med. 2022, 10, 1014. [Google Scholar] [CrossRef]
- Kanlikilicer, P.; Bayraktar, R.; Denizli, M.; Rashed, M.H.; Ivan, C.; Aslan, B.; Mitra, R.; Karagoz, K.; Bayraktar, E.; Zhang, X.; et al. Exosomal MiRNA Confers Chemo Resistance via Targeting Cav1/p-Gp/M2-Type Macrophage Axis in Ovarian Cancer. eBioMedicine 2018, 38, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Torii, C.; Maishi, N.; Kawamoto, T.; Morimoto, M.; Akiyama, K.; Yoshioka, Y.; Minami, T.; Tsumita, T.; Alam, M.T.; Ochiya, T.; et al. MiRNA-1246 in Extracellular Vesicles Secreted from Metastatic Tumor Induces Drug Resistance in Tumor Endothelial Cells. Sci. Rep. 2021, 11, 13502. [Google Scholar] [CrossRef]
- Shao, N.; Song, L.; Sun, X. Exosomal Circ_PIP5K1A Regulates the Progression of Non-Small Cell Lung Cancer and Cisplatin Sensitivity by MiR-101/ABCC1 Axis. Mol. Cell. Biochem. 2021, 476, 2253–2267. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Jiang, H.; Qiao, L.; Guo, C. Circular RNAcirc_0076305 Promotes Cisplatin (DDP) Resistance of Non-Small Cell Lung Cancer Cells by Regulating ABCC1 through MiR-186-5p. Cancer Biother. Radiopharm. 2023, 38, 293–304. [Google Scholar] [CrossRef]
- Pan, Y.; Lin, Y.; Mi, C. Cisplatin-Resistant Osteosarcoma Cell-Derived Exosomes Confer Cisplatin Resistance to Recipient Cells in an Exosomal Circ_103801-Dependent Manner. Cell Biol. Int. 2021, 45, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, H.; Zou, J.; Cao, G.; Li, Y.; Xing, C.; Wu, J. Exosomal Circ_0091741 Promotes Gastric Cancer Cell Autophagy and Chemoresistance via the MiR-330-3p/TRIM14/Dvl2/Wnt/β-Catenin Axis. Hum. Cell 2023, 36, 258–275. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhou, X.; Yin, J.; Zhou, Y. Lnc-PICSAR Contributes to Cisplatin Resistance by MiR-485-5p/REV3L Axis in Cutaneous Squamous Cell Carcinoma. Open Life Sci. 2020, 15, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, L.; Li, J.; Du, Y.; Wang, J.; Liu, J. Effects of Long Noncoding RNA (Linc-VLDLR) Existing in Extracellular Vesicles on the Occurrence and Multidrug Resistance of Esophageal Cancer Cells. Pathol.-Res. Pract. 2019, 215, 470–477. [Google Scholar] [CrossRef]
- Xu, Y.; Qiu, A.; Peng, F.; Tan, X.; Wang, J.; Gong, X. Exosomal Transfer of Circular RNA FBXW7 Ameliorates the Chemoresistance to Oxaliplatin in Colorectal Cancer by Sponging MiR-18b-5p. Neoplasma 2021, 68, 108–118. [Google Scholar] [CrossRef]
- Ji, R.; Zhang, B.; Zhang, X.; Xue, J.; Yuan, X.; Yan, Y.; Wang, M.; Zhu, W.; Qian, H.; Xu, W. Exosomes Derived from Human Mesenchymal Stem Cells Confer Drug Resistance in Gastric Cancer. Cell Cycle 2015, 14, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Ning, K.; Wang, T.; Sun, X.; Zhang, P.; Chen, Y.; Jin, J.; Hua, D. UCH-L1-Containing Exosomes Mediate Chemotherapeutic Resistance Transfer in Breast Cancer. J. Surg. Oncol. 2017, 115, 932–940. [Google Scholar] [CrossRef]
- Ma, X.; Chen, Z.; Hua, D.; He, D.; Wang, L.; Zhang, P.; Wang, J.; Cai, Y.; Gao, C.; Zhang, X.; et al. Essential Role for TrpC5-Containing Extracellular Vesicles in Breast Cancer with Chemotherapeutic Resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 6389–6394. [Google Scholar] [CrossRef]
- Dong, Y.; Pan, Q.; Jiang, L.; Chen, Z.; Zhang, F.; Liu, Y.; Xing, H.; Shi, M.; Li, J.; Li, X.; et al. Tumor Endothelial Expression of P-Glycoprotein upon Microvesicular Transfer of TrpC5 Derived from Adriamycin-Resistant Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2014, 446, 85–90. [Google Scholar] [CrossRef]
- Jigorel, E.; Le Vee, M.; Boursier-Neyret, C.; Parmentier, Y.; Fardel, O. Differential Regulation of Sinusoidal and Canalicular Hepatic Drug Transporter Expression by Xenobiotics Activating Drug-Sensing Receptors in Primary Human Hepatocytes. Drug Metab. Dispos. 2006, 34, 1756–1763. [Google Scholar] [CrossRef] [Green Version]
- Mostafazadeh, M.; Kahroba, H.; Haiaty, S.; TazeKand, A.P.; Samadi, N.; Rahbarghazi, R.; Nouri, M. In Vitro Exosomal Transfer of Nrf2 Led to the Oxaliplatin Resistance in Human Colorectal Cancer LS174T Cells. Cell Biochem. Funct. 2022, 40, 391–402. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Mao, R.; Zhang, Y.; Wen, J.; Liu, Q.; Liu, Y.; Zhang, T. DNAJB8 in Small Extracellular Vesicles Promotes Oxaliplatin Resistance through TP53/MDR1 Pathway in Colon Cancer. Cell Death Dis. 2022, 13, 151. [Google Scholar] [CrossRef]
- Yamashita, M.; Hirohashi, Y.; Torigoe, T.; Kusumoto, H.; Murai, A.; Imagawa, T.; Sato, N. Dnajb8, a Member of the Heat Shock Protein 40 Family Has a Role in the Tumor Initiation and Resistance to Docetaxel but Is Dispensable for Stress Response. PLoS ONE 2016, 11, e0146501. [Google Scholar] [CrossRef]
- Wang, H.; Qi, Y.; Lan, Z.; Liu, Q.; Xu, J.; Zhu, M.; Yang, T.; Shi, R.; Gao, S.; Liang, G. Exosomal PD-L1 Confers Chemoresistance and Promotes Tumorigenic Properties in Esophageal Cancer Cells via Upregulating STAT3/MiR-21. Gene Ther. 2023, 30, 88–100. [Google Scholar] [CrossRef]
- Zhao, W.; Ning, L.; Wang, L.; Ouyang, T.; Qi, L.; Yang, R.; Wu, Y. MiR-21 Inhibition Reverses Doxorubicin-Resistance and Inhibits PC3 Human Prostate Cancer Cells Proliferation. Andrologia 2021, 53, e14016. [Google Scholar] [CrossRef]
- Xie, Z.; Cao, L.; Zhang, J. MiR-21 Modulates Paclitaxel Sensitivity and Hypoxia-Inducible Factor-1α Expression in Human Ovarian Cancer Cells. Oncol. Lett. 2013, 6, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Zhou, Z.-F. Exosomes Derived from Taxol-Resistant Nasopharyngeal Carcinoma (NPC) Cells Transferred DDX53 to NPC Cells and Promoted Cancer Resistance to Taxol. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 127–138. [Google Scholar] [CrossRef]
- Xiao, P.; Liu, Y.-K.; Han, W.; Hu, Y.; Zhang, B.-Y.; Liu, W.-L. Exosomal Delivery of FTO Confers Gefitinib Resistance to Recipient Cells through ABCC10 Regulation in an M6A-Dependent Manner. Mol. Cancer Res. 2021, 19, 726–738. [Google Scholar] [CrossRef]
- Kavanagh, E.L.; Lindsay, S.; Halasz, M.; Gubbins, L.C.; Weiner-Gorzel, K.; Guang, M.H.Z.; McGoldrick, A.; Collins, E.; Henry, M.; Blanco-Fernández, A.; et al. Protein and Chemotherapy Profiling of Extracellular Vesicles Harvested from Therapeutic Induced Senescent Triple Negative Breast Cancer Cells. Oncogenesis 2017, 6, e388. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Luk, F.; Jaiswal, R.; George, A.M.; Grau, G.E.R.; Bebawy, M. Microparticle Drug Sequestration Provides a Parallel Pathway in the Acquisition of Cancer Drug Resistance. Eur. J. Pharmacol. 2013, 721, 116–125. [Google Scholar] [CrossRef]
- Ifergan, I.; Scheffer, G.L.; Assaraf, Y.G. Novel Extracellular Vesicles Mediate an ABCG2-Dependent Anticancer Drug Sequestration and Resistance. Cancer Res. 2005, 65, 10952–10958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goler-Baron, V.; Sladkevich, I.; Assaraf, Y.G. Inhibition of the PI3K-Akt Signaling Pathway Disrupts ABCG2-Rich Extracellular Vesicles and Overcomes Multidrug Resistance in Breast Cancer Cells. Biochem. Pharmacol. 2012, 83, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Dong, C.; Jiang, K.; Sun, R.; Zhou, Y.; Yin, Z.; Lv, J.; Zhang, J.; Wang, Q.; Wang, L. Rab27B Enhances Drug Resistance in Hepatocellular Carcinoma by Promoting Exosome-Mediated Drug Efflux. Carcinogenesis 2020, 41, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Safaei, R.; Larson, B.J.; Cheng, T.C.; Gibson, M.A.; Otani, S.; Naerdemann, W.; Howell, S.B. Abnormal Lysosomal Trafficking and Enhanced Exosomal Export of Cisplatin in Drug-Resistant Human Ovarian Carcinoma Cells. Mol. Cancer Ther. 2005, 4, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebhardt, S.; Ditsch, N.; Nieuwland, R.; Rank, A.; Jeschke, U.; Von Koch, F.; Friese, K.; Toth, B. CEA-, Her2/Neu-, BCRP- and Hsp27-Positive Microparticles in Breast Cancer Patients. Anticancer Res. 2010, 30, 1707–1712. [Google Scholar]
- Saito, J.; Hirota, T.; Furuta, S.; Kobayashi, D.; Takane, H.; Ieiri, I. Association between DNA Methylation in the MiR-328 5′-Flanking Region and Inter-Individual Differences in MiR-328 and BCRP Expression in Human Placenta. PLoS ONE 2013, 8, e72906. [Google Scholar] [CrossRef]
- Gotanda, K.; Hirota, T.; Saito, J.; Fukae, M.; Egashira, Y.; Izumi, N.; Deguchi, M.; Kimura, M.; Matsuki, S.; Irie, S.; et al. Circulating Intestine-Derived Exosomal MiR-328 in Plasma, a Possible Biomarker for Estimating BCRP Function in the Human Intestines. Sci. Rep. 2016, 6, 32299. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Mizutani, K.; Kameyama, K.; Kawakami, K.; Fujita, Y.; Nakane, K.; Kanimoto, Y.; Ehara, H.; Ito, H.; Seishima, M.; et al. Serum Exosomal P-Glycoprotein Is a Potential Marker to Diagnose Docetaxel Resistance and Select a Taxoid for Patients with Prostate Cancer. Urol. Oncol. 2015, 33, 385.e15–385.e20. [Google Scholar] [CrossRef]
- Kharaziha, P.; Chioureas, D.; Rutishauser, D.; Baltatzis, G.; Lennartsson, L.; Fonseca, P.; Azimi, A.; Hultenby, K.; Zubarev, R.; Ullén, A.; et al. Molecular Profiling of Prostate Cancer Derived Exosomes May Reveal a Predictive Signature for Response to Docetaxel. Oncotarget 2015, 6, 21740–21754. [Google Scholar] [CrossRef] [Green Version]
- Wen, G.; Zhou, T.; Gu, W. The Potential of Using Blood Circular RNA as Liquid Biopsy Biomarker for Human Diseases. Protein Cell 2021, 12, 911–946. [Google Scholar] [CrossRef]
- Chen, I.X.; Newcomer, K.; Pauken, K.E.; Juneja, V.R.; Naxerova, K.; Wu, M.W.; Pinter, M.; Sen, D.R.; Singer, M.; Sharpe, A.H.; et al. A Bilateral Tumor Model Identifies Transcriptional Programs Associated with Patient Response to Immune Checkpoint Blockade. Proc. Natl. Acad. Sci. USA 2020, 117, 23684–23694. [Google Scholar] [CrossRef]
- Zemek, R.M.; Fear, V.S.; Forbes, C.; de Jong, E.; Casey, T.H.; Boon, L.; Lassmann, T.; Bosco, A.; Millward, M.J.; Nowak, A.K.; et al. Bilateral Murine Tumor Models for Characterizing the Response to Immune Checkpoint Blockade. Nat. Protoc. 2020, 15, 1628–1648. [Google Scholar] [CrossRef]
- Kim, M.-H.; van Noort, D.; Sung, J.H.; Park, S. Organ-on-a-Chip for Studying Gut-Brain Interaction Mediated by Extracellular Vesicles in the Gut Microenvironment. Int. J. Mol. Sci. 2021, 22, 13513. [Google Scholar] [CrossRef]
- Kim, J.; Lee, C.; Kim, I.; Ro, J.; Kim, J.; Min, Y.; Park, J.; Sunkara, V.; Park, Y.-S.; Michael, I.; et al. Three-Dimensional Human Liver-Chip Emulating Premetastatic Niche Formation by Breast Cancer-Derived Extracellular Vesicles. ACS Nano 2020, 14, 14971–14988. [Google Scholar] [CrossRef]
- Berardocco, M.; Radeghieri, A.; Busatto, S.; Gallorini, M.; Raggi, C.; Gissi, C.; D’Agnano, I.; Bergese, P.; Felsani, A.; Berardi, A.C. RNA-Seq Reveals Distinctive RNA Profiles of Small Extracellular Vesicles from Different Human Liver Cancer Cell Lines. Oncotarget 2017, 8, 82920–82939. [Google Scholar] [CrossRef] [Green Version]
- Njock, M.-S.; O’Grady, T.; Nivelles, O.; Lion, M.; Jacques, S.; Cambier, M.; Herkenne, S.; Muller, F.; Christian, A.; Remacle, C.; et al. Endothelial Extracellular Vesicles Promote Tumour Growth by Tumour-Associated Macrophage Reprogramming. J. Extracell. Vesicles 2022, 11, e12228. [Google Scholar] [CrossRef]
- Yoh, K.E.; Lowe, C.J.; Mahajan, S.; Suttmann, R.; Nguy, T.; Reichelt, M.; Yang, J.; Melendez, R.; Li, Y.; Molinero, L.; et al. Enrichment of Circulating Tumor-Derived Extracellular Vesicles from Human Plasma. J. Immunol. Methods 2021, 490, 112936. [Google Scholar] [CrossRef]
- Ayers, L.; Pink, R.; Carter, D.R.F.; Nieuwland, R. Clinical Requirements for Extracellular Vesicle Assays. J. Extracell. Vesicles 2019, 8, 1593755. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Donor Cell | Target Cell | Relevant EV Cargo | Effect on Target Cell | Reference |
|---|---|---|---|---|
| Drug-resistant MCF7 cells | Drug-sensitive MCF7 cells | P-gp transporter | ↑ P-gp protein expression and activity ↑ Doxorubicin resistance. | [111] |
| Drug-resistant MCF7 cells | Drug-sensitive MCF7 cells | P-gp transporter | ↑ P-gp and MRP1 mRNA expression ↑ Doxorubicin resistance | [112] |
| Docetaxel-resistant MCF7 cells | Drug-sensitive MCF7 cells | P-gp transporter | ↑ P-gp protein expression ↑ Resistance to docetaxel | [113] |
| Drug-resistant MCF7 | Drug-sensitive MCF7 | P-gp mRNA | ↑ P-gp mRNA | [114] |
| Drug-resistant CEM cells | Drug-sensitive CEM cells | P-gp mRNA, miR-326 | ↑ P-gp mRNA ↓ MRP1 mRNA | [114] |
| Drug-resistant CEM cells | Cancerous (drug-sensitive CEM cells) and noncancerous cells (human mammary basal epithelial cells, human osteoblasts, human urothelial cells) | P-gp and MRP1 transporters | ↑ P-gp and MRP1 protein expression | [115] |
| Drug-resistant MCF7 cells | Drug-sensitive MCF7 cells | P-gp transporter, CD44 | ↑ P-gp protein expression | |
| Drug-resistant CEM cells | Drug-sensitive CEM cells | P-gp transporter | ↑ P-gp protein expression ↑ P-gp activity | [117] |
| Vincristine-resistant KB cells | Drug-sensitive KB cells | P-gp transporter | ↑ P-gp protein expression ↑ P-gp activity ↑ Resistance to doxorubicin | [118] |
| Doxorubicin-resistant MG-63 cells | Drug-sensitive MG-63 | P-gp transporter and mRNA | ↑ P-gp mRNA expression ↑ Resistance to doxorubicin | [119,120] |
| Paclitaxel-resistant HGC27 and KATOIII cells lines | Drug-sensitive HGC27 and KATOIII cell lines | P-gp transporter | ↑ Resistance to paclitaxel | [121] |
| Drug-resistant HepG2 cells | Drug-sensitive HepG2 cells, Huh7, SMMC-7721 cells | P-gp transporter | ↑ P-gp protein expression ↑ Resistance to cisplatin | [122] |
| Docetaxel-resistant DU145 and 22Rv1 cells | Drug-sensitive DU145 and 22Rv1 cells | P-gp transporter | ↑ Resistance to docetaxel | [124] |
| Serum EVs from prostate cancer patients who did not respond to chemotherapy | Drug-sensitive DU145 and 22Rv1 cells | ? | ↑ Resistance to docetaxel | |
| MRP1-overexpressing CCRF-CEM cells | CCRF-CEM cells | MRP1 transporter | ↑ MRP1 mRNA expression ↑ MRP1 protein expression ↑ MRP1 activity | [128] |
| MRP1-overexpressing and daunorubicin-resistant HL60/AR cells | Drug-sensitive HL60 cells | MRP1 transporter | ↑ Resistance to daunorubicin ↑ MRP1 activity | [129] |
| Plasma EVs from patients with newly diagnosed and recurrent AML | U937 cells | ? | ↑ Resistance to idarubicin ↑ P-gp and MRP1 mRNA expression | [132] |
| Cisplatin-resistant MDA-MB-231 cells | MDA-MB-231, MCF7, SKBR3 cells | miR-423-5p | ↑ P-gp expression ↑ Resistance to cisplatin | [139] |
| MDA-MB-231 cells treated with docetaxel or doxorubicin | MDA-MB-231 cells | miR-9-5p, miR-195-5p, miR-203a-3p | ↑ P-gp, MRP1, and BCRP expression | [140] |
| Drug-resistant SKOV3 cells | Drug-sensitive A2780 cells | miR-429 | ↑ Resistance to cisplatin | [142] |
| Drug-resistant Bel7402 cells | Drug-sensitive Bel7402 cells | miR-32-5p | ↑ Resistance to 5-FU, oxaliplatin, sorafenib, and gemcitabine | [143] |
| MSC | Cisplatin- and vincristine-resistant SGC7901 cells | miR-301b-3p | ↑ P-gp expression | [144] |
| MSC | MPC-11 cells | miR-155 | ↑ P-gp, MRP1, and BCRP expression ↑ Resistance to bortezomib and dexamethasone | [145] |
| EVs from M2 tumor-associated macrophages (M2) | A2780 cells | miR-221-3p | ↑ P-gp expression (in the presence of cisplatin) ↑ MRP1 and BCRP expression | [146] |
| Cisplatin-resistant A2780 cells (A2780/DDP) | ↑ P-gp expression | |||
| SKOV3-ip1 cells | M2 macrophages | miR-1246 | ↑ Caveolin-1 mRNA expression | [147] |
| A375SM cells | Tumor endothelial cells | miR-1246 | ↑ Resistance to 5-FU | [148] |
| A549 and H1299 cells | A549 and H1299 cells | circ_PIP5K1A (hsa_circ_0014130) | ↑ Resistance to cisplatin | [149] |
| A549 and H1299 cells | A549 and H1299 cells | circ_0076305 | ↑ MRP1 expression ↑ Resistance to cisplatin | [150] |
| Cisplatin-resistant MG-63/CDDP cells | Drug-sensitive MG-63 and U2OS cells | hsa_circ_103801 | ↑ Resistance to cisplatin ↑ P-gp and MRP1 expression | [151] |
| Oxaliplatin-resistant HGC27 cells | Drug-sensitive HGC27 cells | hsa_circ_0091741 | ↑ P-gp, MRP1, and LRP1 expression | [152] |
| Cisplatin-resistant HSC-5 cells | Drug-sensitive HSC-5 cells | lnc PICSAR | ↑ P-gp and MRP1 expression | [153] |
| Eca109 cells (treated with doxorubicin) | Eca109 cells (untreated) | lncRNA VLDLR | ↑ Resistance to doxorubicin | [154] |
| MSC | Gastric cancer cells | ? | ↑ P-gp, MRP1, and LRP expression ↑ Resistance to 5-FU | [156] |
| Doxorubicin-resistant MCF7 cells | Drug-sensitive MCF7 cells | UCH-L1 | ↑ P-gp expression ↑ Resistance to doxorubicin | [157] |
| Doxorubicin-resistant MCF7 cells | Drug-sensitive MCF7 cells | TrpC5 and P-gp mRNA | ↑ P-gp expression ↑ Resistance to doxorubicin | [158] |
| Doxorubicin-resistant MCF7 cells | HMECs | TrpC5 | ↑ P-gp expression | [159] |
| Oxaliplatin-resistant LS174T cells | Drug-sensitive LS174T cells | Nrf2 | ↑ P-gp expression ↑ P-gp activity ↑ Resistance to oxaliplatin | [161] |
| Oxaliplatin-resistant SW480 and SW620 cells | Drug-sensitive SW480 and SW620 cells | DNAJB8 | ↑ P-gp expression ↑ Resistance to oxaliplatin | [162] |
| Paclitaxel-resistant EC-9706 cells | Drug-sensitive EC-9706 cells | PD-L1 | ↑ P-gp expression ↑ Resistance to paclitaxel | [164] |
| Paclitaxel-resistant CNE1 cells | Drug-sensitive CNE1 cells | DDX53 | ↑ P-gp expression ↑ Resistance to paclitaxel | [167] |
| Gefitinib-resistant PC9 and H1975 cells | Drug-sensitive PC9 cells | FTO | ↑ BCRP and ABCC10 expression ↑ Resistance to gefitinib | [168] |
| Donor Cell | Target Cell | Relevant EV Cargo | Effect on the Target Cell | Reference |
|---|---|---|---|---|
| P-gp-overexpressing CEM cells | MRP1-overexpressing CEM cells | miR-326 | ↓ MRP1 mRNA and protein expression | [133] |
| Drug-sensitive SGC-7901 and MGC-803 cells | 5-FU/cisplatin-resistant SGC-7901 cells | miR-107 | ↓ P-gp expression ↑ Sensitivity to 5-FU and cisplatin | [134] |
| Parental MG-63 cells (treated with luteolin) | Doxorubicin-resistant MG-63 cells | miR-384 | ↓ P-gp expression ↑ Sensitivity to doxorubicin | [135] |
| Drug-sensitive MCF7 cells (treated with β-elemene) | Doxorubicin- and docetaxel-resistant MCF7 cells | ↑ miR-34 and ↓ miR-452 | ↓ P-gp expression | [136] |
| hCECs (nontumoral cells) | HepG2 and Hep3B cells | miR-214 | ↓ P-gp expression ↑ Sensitivity to oxaliplatin and sorafenib | [137] |
| MSCs | U87 and T98G cells | anti-miR-9 | ↓ P-gp expression | [138] |
| Oxaliplatin-resistant SW480 and HCT116 cells | Drug-sensitive SW480 and HCT116 cells | circ_FBXW7 | ↑ Sensitivity to oxaliplatin ↓ miR-18b-5p and MRP1 expression | [155] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bucci-Muñoz, M.; Gola, A.M.; Rigalli, J.P.; Ceballos, M.P.; Ruiz, M.L. Extracellular Vesicles and Cancer Multidrug Resistance: Undesirable Intercellular Messengers? Life 2023, 13, 1633. https://doi.org/10.3390/life13081633
Bucci-Muñoz M, Gola AM, Rigalli JP, Ceballos MP, Ruiz ML. Extracellular Vesicles and Cancer Multidrug Resistance: Undesirable Intercellular Messengers? Life. 2023; 13(8):1633. https://doi.org/10.3390/life13081633
Chicago/Turabian StyleBucci-Muñoz, María, Aldana Magalí Gola, Juan Pablo Rigalli, María Paula Ceballos, and María Laura Ruiz. 2023. "Extracellular Vesicles and Cancer Multidrug Resistance: Undesirable Intercellular Messengers?" Life 13, no. 8: 1633. https://doi.org/10.3390/life13081633
APA StyleBucci-Muñoz, M., Gola, A. M., Rigalli, J. P., Ceballos, M. P., & Ruiz, M. L. (2023). Extracellular Vesicles and Cancer Multidrug Resistance: Undesirable Intercellular Messengers? Life, 13(8), 1633. https://doi.org/10.3390/life13081633