Chemical Transformations in Proto-Cytoplasmic Media. Phosphorus Coupling in the Silica Hydrogel Phase


Abstract
:1. Introduction
2. Materials and Methods
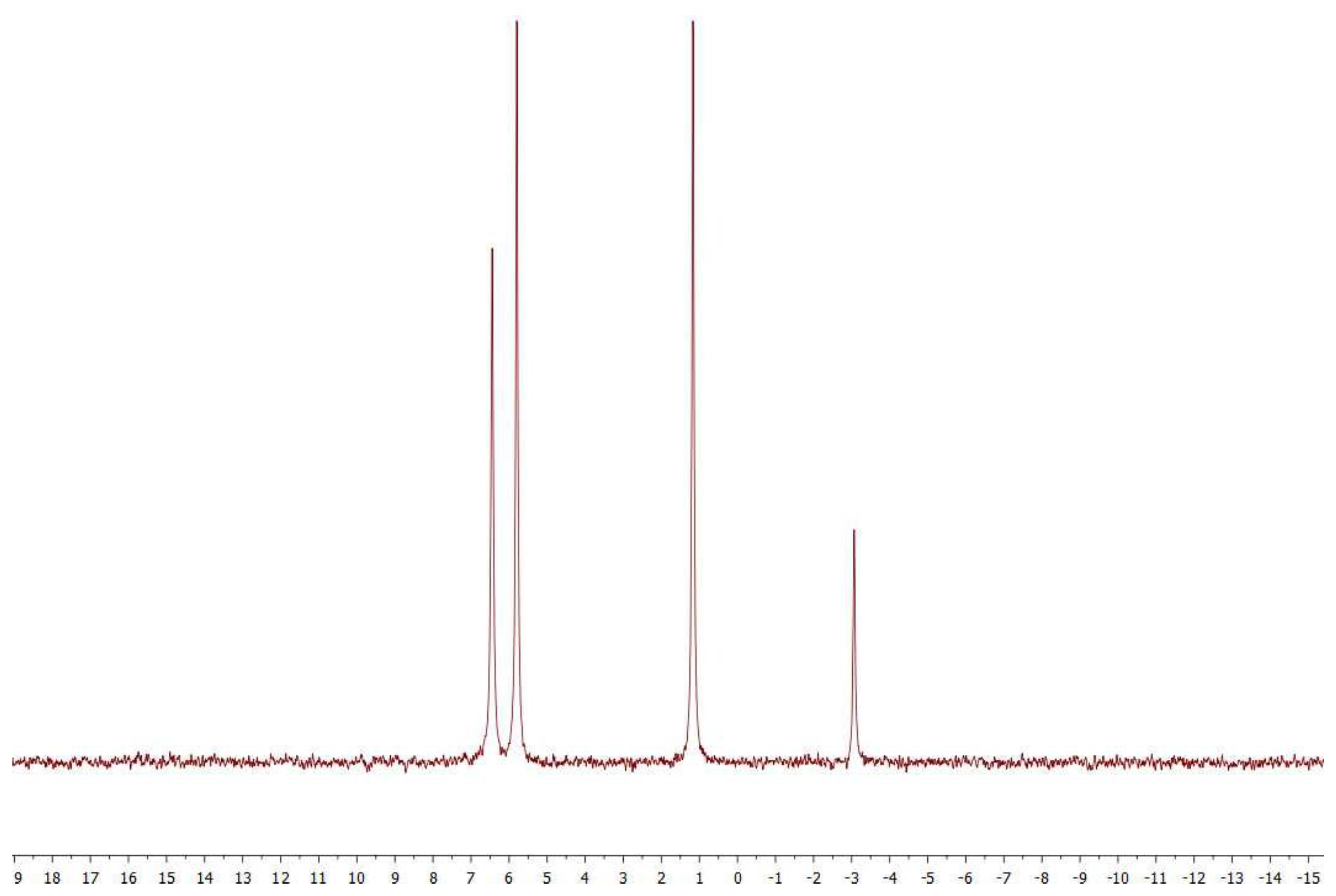
2.1. Analytical Methods
2.2. Preparation of Silica Hydrogels (SHGs)
2.3. General Methods for Preparing & Analyzing Phosphorus-Implanted SHGs
2.4. Control Experiments in Aqueous Solution
- C1: H2O (6.6 mL), Pi(III) (0.5 M), Pi(V) (0.5 M), Fe(II) (100 mg), glacial acetic acid (200 μL)
- C2: H2O (6.6 mL), PPi(III– V) (0.5 M), Fe(II) (100 mg), glacial acetic acid (200 μL)
- C3: H2O (6.6 mL), PPi(III– V) (0.5 M), Fe(II) (100 mg), glacial acetic acid (200 μL), H2O2 (1 mL, 0.2 M)
- C4: H2O (6.6 mL), Pi(III) (0.5 M), Pi(V) (0.5 M), Fe(II) (100 mg), glacial acetic acid (200 μL), H2O2 (1 mL, 0.2 M)
- C5: H2O (6.6 mL), Pi(III) (0.5 M), Pi(V) (0.5 M), Fe(II) (100 mg), glacial acetic acid (200 μL), silica gel GF254 (0.2 g)
- C6: H2O (6.6 mL), PPi(III– V) (0.5 M), Fe(II) (100 mg), glacial acetic acid (200 μL), silica gel GF254 (0.2 g).
3. Results
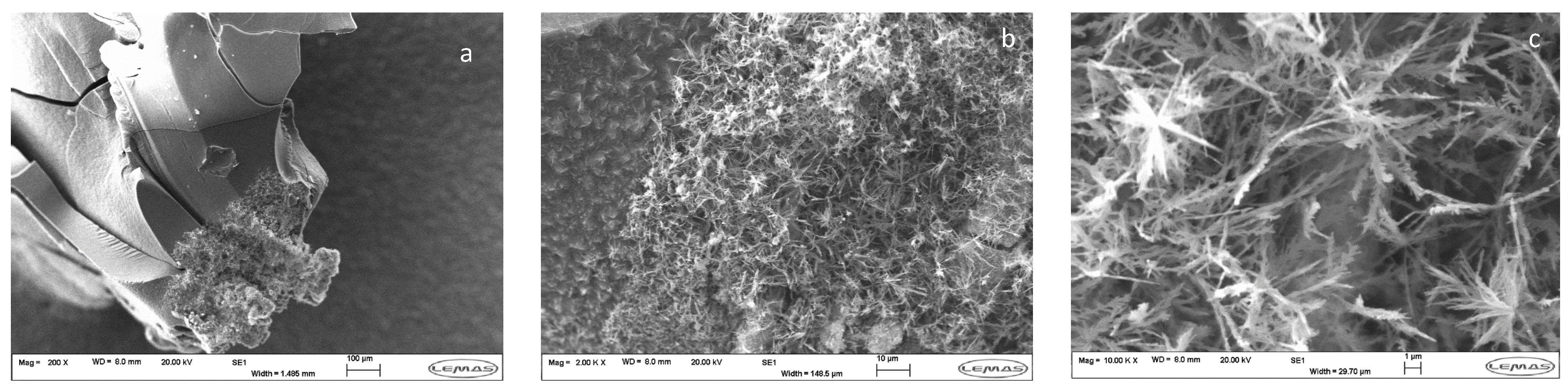
3.1. Preparation and Analysis of SHGs
3.2. Coupling of Pi within the Silica Hydrogel Phase
- (1)
- No Pi coupling was observed under any conditions where hydrochloric acid (HCl) was used as the low pH component of the gel system.
- (2)
- No metal additive, other than Fe(II), afforded any Pi coupling.
- (3)
- Successful Pi coupling protocols using acetic acid also delivered positive results for Pi coupling when acetic was replaced by formic acid.
- (4)
- No successful Pi coupling was observed when the Pi components were used seperately, Pi(III) or Pi(V).
- (5)
- Successful Pi coupling was achieved only using a 1:1 mixture of Pi(III) and Pi(V) or pure PPi(III– V).
- (6)
- Successful Pi coupling was observed both when the Fe(II) additive used was employed either in solution or as a heterogeneous addition to the pre-formed gel.
- (7)
- Successful Pi coupling was observed with Fe(II) only when formulated aerobically. No coupling was observed under anaerobic conditions.
- (8)
- Control experiments performed under aqueous (non-gelled) conditions revealed no Pi coupling but distinct oxidation of PPi(III– V) in the presence of Fe(II)-air [1.3% conversion of PPi(III– V) to PPi(V)] and pronounced oxidation in the presence of the Fenton system, Fe(II)-H2O2 [35.1% conversion of PPi(III– V) to PPi(V)].
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fitz, D.; Reiner, H.; Rode, B.M. Chemical evolution toward the origin of life. Pure Appl. Chem. 2007, 79, 2101–2117. [Google Scholar] [CrossRef]
- Danger, G.; Plasson, R.; Pascal, R. Pathways for the formation and evolution of peptides in prebiotic environments. Chem. Soc. Rev. 2012, 41, 5416–5429. [Google Scholar] [CrossRef] [PubMed]
- Danger, G.; Boiteau, L.; Cottet, H.; Pascal, R. The peptide formation mediated by cyanate revisited. N-Carboxyanhydrides as accessible intermediates in the decomposition of N-carbamoylamino acids. J. Am. Chem. Soc. 2006, 128, 7412–7413. [Google Scholar] [CrossRef] [PubMed]
- Leman, L.; Orgel, L.; Ghadiri, M.R. Carbonyl sulfide-mediated prebiotic formation of peptide. Science 2004, 306, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Bujdak, J.; Rode, B.M. Preferential amino acid sequences in alumina-catalyzed peptide bond formation. J. Inorg. Biochem. 2002, 90, 1–7. [Google Scholar] [CrossRef]
- Le Son, H.; Suwannachot, Y.; Bujdak, J.; Rode, B.M. Salt-induced peptide formation from amino acids in the presence of clays and related catalysts. Inorg. Chim. Acta 1998, 272, 89–94. [Google Scholar] [CrossRef]
- Forsythe, J.G.; Yu, S.-S.; Mamajanov, I.; Grover, M.A.; Krishnamurthy, R.; Fernandez, F.M.; Hud, N. Ester-Mediated Amide Bond Formation Driven by Wet-Dry Cycles: A Possible Path to Polypeptides on the Prebiotic Earth. Angew. Chem. Int. Ed. 2015, 54, 9871–9875. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, M.; Surman, A.J.; Cooper, G.J.T.; Suarez-Marina, I.; Hosni, Z.; Lee, M.P.; Cronin, L. Formation of oligopeptides in high yield under simple programmable conditions. Nat. Commun. 2015, 6, 8385. [Google Scholar] [CrossRef] [PubMed]
- Cafferty, B.J.; Fialho, D.M.; Khanam, J.K.; Krishnamurthy, R.; Hud, N.V. Spontaneous formation and base pairing of plausible prebiotic nucleotides in water. Nat. Commun. 2016, 7, 11328. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F. In silico Support for Eschenmoser’s Glyoxylate Scenario. Israel J. Chem. 2015, 55, 919–933. [Google Scholar] [CrossRef]
- Bowler, F.R.; Chan, C.K.W.; Duffy, C.D.; Gerland, B.; Islam, S.; Powner, M.W.; Sutherland, J.D.; Xu, J. Prebiotically plausible oligoribonucleotide ligation facilitated by chemoselective acetylation Common origins of RNA, protein and lipid precursors in a cyanosulfidic protometabolism. Nat. Chem. 2013, 5, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.H.; Percivalle, C.; Ritson, D.J.; Duffy, C.D.; Sutherland, J.D. Common origins of RNA, protein and lipid precursors in a cyanosulfidic protometabolism. Nat. Chem. 2015, 7, 301–307. [Google Scholar]
- Hernandez, A.R.; Piccirilli, J.A. Chemical origins of life. Prebiotic RNA unstuck. Nat. Chem. 2013, 5, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Cozens, C.; Mutschler, H.; Nelson, G.M.; Houlihan, G.; Taylor, A.I.; Holliger, P. Enzymatic Synthesis of Nucleic Acids with Defined Regioisomeric 2′–5′ Linkages. Angew. Chem. Int. Ed. Engl. 2015, 54, 15570–15573. [Google Scholar] [CrossRef] [PubMed]
- Yeates, J.A.M.; Hilbe, C.; Zwick, M.; Nowak, M.A.; Lehman, N. Dynamics of prebiotic RNA reproduction illuminated by chemical game theory. Proc. Natl. Acad. Sci. USA 2016, 113, 5030–5035. [Google Scholar] [CrossRef] [PubMed]
- Zwicker, D.; Seyboldt, R.; Weber, C.A.; Hyman, A.A.; Juelicher, F. Growth and division of active droplets: A model for protocells. Growth Phys. 2016, 1–28. [Google Scholar] [CrossRef]
- Arai, N.; Yoshimoto, Y.; Yasuoka, K.; Ebisuzaki, T. Self-assembly behaviours of primitive and modern lipid membrane solutions: a coarse-grained molecular simulation study. Phys. Chem. Chem. Phys. 2016, 18, 19426–19432. [Google Scholar] [CrossRef] [PubMed]
- Hanczyc, M.M.; Monnard, P.-A. The origin of life and the potential role of soaps. Lipid Technol. 2016, 28, 88–92. [Google Scholar] [CrossRef]
- Saha, R.; Chen, I.A. Origin of Life: Protocells Red in Tooth and Claw. Curr. Biol. 2015, 25, R1175–R1177. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.Z.; Hentrich, C.; Szostak, J.W. Rapid RNA Exchange in Aqueous Two-Phase System and Coacervate Droplets. Orig. Life Evol. Biosph. 2014, 44, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulkidjanian, A.Y.; Makarova, K.S.; Galperin, M.Y.; Koonin, E.V. Inventing the dynamo machine: The evolution of the F-type and V-type ATPases. Nat. Rev. Microbiol. 2007, 5, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Herschy, B.; Whicher, A.; Camprubi, E.; Watson, C.; Dartnell, L.; Ward, J.; Evans, J.R.G.; Lane, N. An Origin-of Life Reactor to Simulate Alkaline Hydrothermal Vents. J. Mol. Evol. 2014, 79, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Barge, L.M.; Kee, T.P.; Doloboff, I.J.; Hampton, J.M.P.; Ismail, M.; Pourkashanian, M.; Zeytounian, J.; Baum, M.M.; Moss, J.A.; Lin, C.-K.; et al. The Fuel Cell Model of Abiogenesis: A New Approach to Origin-of-Life Simulations. Astrobiology 2014, 14, 254–270. [Google Scholar] [CrossRef] [PubMed]
- Barge, L.M.; Doloboff, I.J.; Russell, M.J.; Vander Velde, D.; White, L.M.; Stucky, G.D.; Baum, M.M.; Zeytounian, J.; Kidd, R.; Kanik, I. Pyrophosphate synthesis in iron mineral films and membranes simulating prebiotic submarine hydrothermal precipitates. Geochim. Cosmochim. Acta 2014, 128, 1–12. [Google Scholar] [CrossRef]
- Barge, L.M.; Branscomb, E.; Brucato, J.R.; Cardoso, S.S.; Cartwright, J.H.E.; Danielache, O.; Galante, D.; Kee, T.P.; Miguel, Y.; Mojzsis, S.; et al. Thermodynamics, Disequilibrium, Evolution: Far-From-Equilibrium Geological and Chemical Considerations for Origin-Of-Life Research. Orig. Life Evol. Biosph. 2017, 47, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, A.; Burkhardt, J.; Cockell, C.S.; Cray, J.A.; Dijksterhuis, J.; Fox-Powell, M.; Kee, T.P.; Kminek, G.; McGenity, T.J.; Timmis, K.N.; et al. Multiplication of microbes below 0.690 water activity: Implications for terrestrial and extraterrestrial life. Environ. Microbiol. 2015, 17, 257–277. [Google Scholar] [CrossRef] [PubMed]
- Cape, J.L.; Monnard, P.-A.; Boncella, J.M. Prebiotically relevant mixed fatty acid vesicles support anionic solute encapsulation and photochemically catalyzed trans-membrane charge transport. Chem. Sci. 2011, 2, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Georgelin, T.; Jaber, M.; Bazzi, H.; Lambert, J.-F. Formation of Activated Biomolecules by Condensation on Mineral Surfaces—A Comparison of Peptide Bond Formation and Phosphate Condensation. Orig. Life Evol. Biosph. 2014, 43, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Mutschler, H.; Wochner, A.; Holliger, P. Freeze-thaw cycles as drivers of complex ribozyme assembly. Nat. Chem. 2015, 7, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Dass, A.V.; Hickman-Lewis, K.; Brack, A.; Kee, T.P.; Westall, F. Stochastic prebiotic chemistry within realistic geological systems. ChemistrySelect 2016, 1, 4906–4926. [Google Scholar] [CrossRef]
- Trevors, J.T.; Gerald, H.; Pollack, G.H. Hypothesis: The origin of life in a hydrogel environment. Prog. Biophys. Mol. Biol. 2005, 89, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Trevors, J.T. Perspective: Researching the transition from non-living to the first microorganisms: Methods and experiments are major challenges. J. Microbiol. Methods 2010, 81, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Trevors, J.T. Origin of microbial life: Nano- and molecular events, thermodynamics/entropy, quantum mechanisms and genetic instructions. J. Microbiol. Methods 2011, 84, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Trevors, J.T. Hypothesized origin of microbial life in a prebiotic gel and the transition to a living biofilm and microbial mats. Compets Rendus Biol. 2011, 334, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Pollack, G.H. Cells, Gels and the Engines of Life; Ebner and Sons: Seattle, WA, USA, 2001; ISBN 13:978-0962689529. [Google Scholar]
- Yang, D.; Peng, S.; Hartman, M.R.; Gupton-Campolongo, T.; Rice, E.J.; Chang, A.K.; Gu, Z.; Lu, G.Q.; Luo, D. Enhanced transcription and translation in clay hydrogel and implications for early life evolution. Sci. Rep. 2013, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.C.; Sousa, F.L.; Mrnjavac, N.; Neukirchen, S.; Roettger, M.; Nelson-Sathi, S.; Martin, W.F. The physiology and habitat of the last universal common ancestor. Nat. Microbiol. 2016, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kee, T.P.; Monnard, P.-A. On the Emergence of a Proto-Metabolism and the Assembly of Early Protocells. Elements 2016, 12, 419–424. [Google Scholar] [CrossRef]
- Santiburcio, D.M.; Marx, D. Chemistry in nano-confined water. Chem. Sci. 2017, 8, 3444–3452. [Google Scholar] [CrossRef] [PubMed]
- Hansma, H.G. Better than Membranes at the Origin of Life? Life 2017, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.; Pohorille, A.; Chen, I.A. Molecular Crowding and Early Evolution. Orig. Life Evol. Biosph. 2014, 44, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Oparin, A.I.; Serebrovskaia, K.B.; Vasileva, N.V.; Balaevsk, T.O. Formation of coacervates from polypeptides and polynucleotides. Dokl. Akad. Nauk. 1961, 154, 407–412. [Google Scholar]
- Fox, W. The evolutionary significance of phase-separated microsystems. Orig. Life Evol. Biosph. 1976, 7, 49–68. [Google Scholar] [CrossRef]
- Mann, S. The origins of life: Old problems, new chemistries. Angew. Chem. 2013, 52, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Copley, S.D.; Smith, E.; Morowitz, H.J. The origin of RNA world: Co-evolution of genes and metabolism. Bioorg. Chem. 2007, 35, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Grenne, T.; Slack, J.F. Bedded jaspers of the Ordovician Løkken ophiolite, Norway: Seafloor deposition and diagenetic maturation of hydrothermal plume-derived silica-iron gels. Miner. Deposita 2003, 38, 625–639. [Google Scholar] [CrossRef]
- Papineau, D. Mineral Environments on the Early Earth. Elements 2010, 6, 25–30. [Google Scholar] [CrossRef]
- Kirkpatrick, J.D.; Rowe, C.D.; White, J.C.; Brodsky, E.E. Silica gel formation during fault slip: Evidence from the rock record. Geology 2013. [Google Scholar] [CrossRef]
- Serrano, A.; Perez-Castineira, J.R.; Baltscheffsky, H.; Baltscheffsky, M. Proton-Pumping Inorganic Pyrophosphatases in Some Archaea and Other Extremophilic Prokaryotes. J. Bioenergy Biomembr. 2004, 36, 127–133. [Google Scholar] [CrossRef]
- Serrano, A.; Perez-Castineira, J.R.; Baltscheffsky, M.; Baltscheffsky, H. H+-PPases: Yesterday, today and tomorrow. Int. Union Biochem. Mol. Biol. 2007, 59, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Baltscheffsky, M.; Schultz, A.; Baltscheffsky, H. H+-proton-pumping inorganic pyrophosphatase: A tightly membrane-bound family. FEBS Lett. 1999, 452, 121–127. [Google Scholar] [CrossRef]
- Feng, N.; Sun, S.; Chao, H.; Zhao, Y. N-phosphorylation of amino acids by trimetaphosphate in aqueous solution—Learning from prebiotic synthesis. Green Chem. 2009, 11, 569–573. [Google Scholar]
- Cheng, C.; Fan, C.; Wan, R.; Tong, C.; Miao, Z.; Chen, J.; Zhao, Y. Phosphorylation of adenosine with trimetaphosphate under simulated prebiotic conditions. Orig. Life Evol. Biosph. 2002, 32, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Nemoto, A.; Imai, E.-I.; Honda, H.; Hatori, K.; Matsuno, K. Phosphorylation of nucleotide molecules in hydrothermal environments. Orig. Life Evol. Biosph. 2004, 34, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.E.; Greenfield, D.; Walshaw, R.D.; Johnson, B.R.G.; Herschy, B.; Smith, C.; Pasek, M.A.; Telford, R.; Scowen, I.; Munshi, T.; et al. Hydrothermal modification of the Sikhote-Alin iron meteorite under low pH geothermal environments. A plausibly prebiotic route to activated phosphorus on the early Earth. Geochim. Cosmochim. Acta 2013, 109, 90–112. [Google Scholar] [CrossRef]
- Bryant, D.E.; Herschy, B.; Marriott, K.E.R.; Cosgrove, N.E.; Pasek, M.A.; Atlas, Z.D.; Cousins, C.R.; Kee, T.P. Phosphate Activation via Reduced Oxidation State Phosphorus (P). Mild Routes to Condensed-P Energy Currency Molecules. Life 2013, 3, 386–402. [Google Scholar] [Green Version]
- Kaye, K.; Bryant, D.E.; Marriott, K.E.R.; Fishwick, C.W.G.; Kee, T.P. Selective phosphonylation of 5′-adenosine monophosphate (5′-AMP) via pyrophosphate [PPi(III)]. Orig. Life Evol. Biosph. 2016, 46, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Madhurambal, G.; Subha, R.; Mojumdar, S.C. Crystallization and thermal characterization of calcium hydrogen phosphate dihydrate crystals. J. Therm. Anal. Calorim. 2009, 96, 73–76. [Google Scholar] [CrossRef]
- Steinman, G.; Kenyon, D.H.; Calvin, M. Dehydration condensation in aqueous solution. Nature 1965, 206, 707–708. [Google Scholar] [CrossRef]
- Pasek, M.A.; Kee, T.P. On the Origin of Phosphorylated Biomolecules. In Origins of Life: The Primal Self-Organization; Egel, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Barge, L.; Hammond, D.; Chan, M.; Potter, S.; Petruska, J.; Nealson, K. Precipitation patterns formed by self-organizing processes in porous media. Geofluids 2011, 11, 124–133. [Google Scholar] [CrossRef]
- ASTM D1141: Standard Specification for Substitute Ocean Water. Available online: https://global.ihs.com/doc_detail.cfm?&input_doc_number=&input_doc_title=&document_name=ASTM%20D1141&item_s_key=00015449&item_key_date=070131&origin=DSSC (accessed on 3 November 2017).
- Ahmed, E.M. Hydrogel: Preparation, characterization, and applications: A review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Cammarata, M.; Caronna, C.; Fiandaca, G.; Levantino, M.; Schir, G.; Vitrano, E.; Cupane, A. Water and Proteins confined in Silica Hydrogels and Silica Nanoparticles: Structural, Dynamic and Functional Studies. In Proceedings of theProgress in Condensed Matter Physics: Festschrift in Honour of Vincenzo Grasso, Messina, Mexico, 9 January 2003; pp. 139–152. [Google Scholar]
- Hu, X.; Wang, X.; Liu, J.; Zhang, S.; Jiang, C.; He, X. Fabrication of mesoporous dendritic silica nanofibers by using dendritic polyaniline templates. Mater. Chem. Phys. 2012, 137, 17–21. [Google Scholar] [CrossRef]
- Alkimim, I.P.; Silva, L.L.; Cardoso, D. Synthesis of hybrid spherical silicas and application in catalytic transesterification reaction. Microporous Mesoporous Mater. 2017. [Google Scholar] [CrossRef]
- Christy, A.A. Quantitative determination of surface area of silica gel particles by near infrared spectroscopy and chemometrics. Physicochem. Eng. Asp. 2008, 3221, 248–252. [Google Scholar] [CrossRef]
- Pasek, M.A.; Kee, T.P.; Bryant, D.E.; Pavlov, A.A.; Lunine, J.L. Production of Potentially Prebiotic Condensed Phosphates by Phosphorus Redox Chemistry. Angew. Chem. Int. Ed. 2008, 47, 7918–7920. [Google Scholar] [CrossRef] [PubMed]
- Mistry, D.; Powles, N. The relative hydrolytic reactivities of pyrophosphites and pyrophosphates. Org. Biomol. Chem. 2013, 11, 5727–5733. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.P.; Banwart, S.A. A geochemical model for removal of iron(II)(aq) from mine water discharges. Appl. Geochem. 2002, 17, 431–443. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent Systems | Water | Standard mean ocean water (SMOW) | ||
| Phosphorus (Pi) Components | 1:1 molar Pi(V) & Pi(III) (0.5 M each) | Pi(III) (0.5 M) | Pi(V) (0.5 M) | PPi(III– V) (0.5 M) |
| Metal Additives | Fe(II) | Fe(III) | Cu(II) | Mg(II) |
| Acid Components | Acetic acid | Formic acid | Hydrochloric acid |
| Sample | Solvent | Acid | Pi Compounds | Metal Additives | Additive Delivery | Mass (g) | PPi(V) (%) | |
|---|---|---|---|---|---|---|---|---|
| G1 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Fe(II) | Deposited | 25 | 0.30 | a |
| G2 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Fe(II) | Deposited | 25 | 0.47 | b |
| G3 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Fe(II) | Solution | 25 | 0.10 | c |
| G4 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Fe(II) | Deposited | 25 | 0.21 | |
| G5 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Fe(II) | Deposited | 25 | 0.10 | |
| G6 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Fe(II) | Deposited | 25 | 0.23 | d |
| G7 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Fe(II) | Solution | 250 | 0.66 | |
| G8 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Fe(II) | Deposited | 250 | 0.40 | |
| G9 | H2O | HCO2H | 1:1 PPi(III)-Pi(V) | Fe(II) | Deposited | 100 | 0.13 | |
| G10 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Fe(II) | Deposited | 100 | 0.55 | e |
| G11 | H2O | MeCO2H | PPi(III– V) | Fe(II) | Deposited | 100 | 3.2 ± 1.0 | f |
| G12 | H2O | MeCO2H | PPi(III– V) | Fe(II) | Deposited | 100 | 2.4 ± 1.0 | f |
| G13 | H2O | MeCO2H | PPi(III– V) | Fe(II) | Deposited | 100 | 12.6 ± 0.5 | g |
| G14 | H2O | MeCO2H | PPi(III– V) | Fe(II) | Deposited | 100 | 9.3 ± 0.5 | h |
| G15 | H2O | MeCO2H | PPi(III– V) | Fe(II) | Deposited | 100 | 5.7 ± 0.6 | i |
| G16 | H2O | MeCO2H | PPi(III– V) | Fe(II) | Solution | 100 | 20 ± 5.0 | g |
| G17 | H2O | MeCO2H | PPi(III– V) | Fe(II) | Solution | 100 | 15.6 ± 3.0 | h |
| G18 | H2O | MeCO2H | PPi(III– V) | Fe(II) | Solution | 100 | 10.7 ± 0.6 | i |
| G19 | H2O | MeCO2H | PPi(III– V) | None | None | 0 | 1.8 ± 0.4 | j |
| G20 | H2O | MeCO2H | PPi(III– V) | None | None | 0 | 1.8 ± 0.2 | k |
| G21 | H2O | MeCO2H | PPi(III– V) | None | None | 0 | 1.3 ± 0.5 | l |
| G22 | H2O | MeCO2H | PPi(III– V) | Fe(II) | Deposited | 100 | N/O | m |
| G23 | H2O | HCl | 1:1 PPi(III)-Pi(V) | Fe(II) | Deposited | 100 | N/O | |
| G24 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Fe(III) | Solution | 250 | N/O | |
| G25 | H2O | MeCO2H | Pi(III) | Fe(III) | Solution | 250 | N/O | |
| G26 | H2O | MeCO2H | Pi(V) | Fe(III) | Solution | 250 | N/O | |
| G27 | H2O | MeCO2H | Pi(V) | Fe(III) | Deposited | 25 | N/O | |
| G28 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Fe(III) | Deposited | 25 | N/O | |
| G29 | H2O | MeCO2H | Pi(III) | Fe(III) | Deposited | 25 | N/O | |
| G30 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Cu(II) | Deposited | 25 | N/O | |
| G31 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Cu(II) | Deposited | 100 | N/O | |
| G32 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Cu(II) | Deposited | 250 | N/O | |
| G33 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Mg(II) | Deposited | 25 | N/O | |
| G34 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Mg(II) | Deposited | 100 | N/O | |
| G35 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Mg(II) | Deposited | 250 | N/O | |
| G36 | SMOW | MeCO2H | 1:1 PPi(III)-Pi(V) | Fe(II) | Deposited | 250 | 0.80 | n |
| C1 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Fe(II) | Solution | 100 | N/O | o |
| C2 | H2O | MeCO2H | PPi(III– V) | Fe(II) | Solution | 100 | 1.3 ± 0.5 | o |
| C3 | H2O | MeCO2H | PPi(III– V) | Fe(II) | Solution | 100 | 35.1 ± 5.0 | p |
| C4 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Fe(II) | Solution | 100 | N/O | p |
| C5 | H2O | MeCO2H | 1:1 PPi(III)-Pi(V) | Fe(II) | Solution | 100 | N/O | q |
| C6 | H2O | MeCO2H | PPi(III– V) | Fe(II) | Solution | 100 | 1.2 ± 0.5 | q |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorrell, I.B.; Henderson, T.W.; Albdeery, K.; Savage, P.M.; Kee, T.P. Chemical Transformations in Proto-Cytoplasmic Media. Phosphorus Coupling in the Silica Hydrogel Phase. Life 2017, 7, 45. https://doi.org/10.3390/life7040045
Gorrell IB, Henderson TW, Albdeery K, Savage PM, Kee TP. Chemical Transformations in Proto-Cytoplasmic Media. Phosphorus Coupling in the Silica Hydrogel Phase. Life. 2017; 7(4):45. https://doi.org/10.3390/life7040045
Chicago/Turabian StyleGorrell, Ian B., Timothy W. Henderson, Kamal Albdeery, Philip M. Savage, and Terence P. Kee. 2017. "Chemical Transformations in Proto-Cytoplasmic Media. Phosphorus Coupling in the Silica Hydrogel Phase" Life 7, no. 4: 45. https://doi.org/10.3390/life7040045
APA StyleGorrell, I. B., Henderson, T. W., Albdeery, K., Savage, P. M., & Kee, T. P. (2017). Chemical Transformations in Proto-Cytoplasmic Media. Phosphorus Coupling in the Silica Hydrogel Phase. Life, 7(4), 45. https://doi.org/10.3390/life7040045