Chemistry of Homocysteine Thiolactone in A Prebiotic Perspective



Abstract
:
{kind=link}
{kind=link}
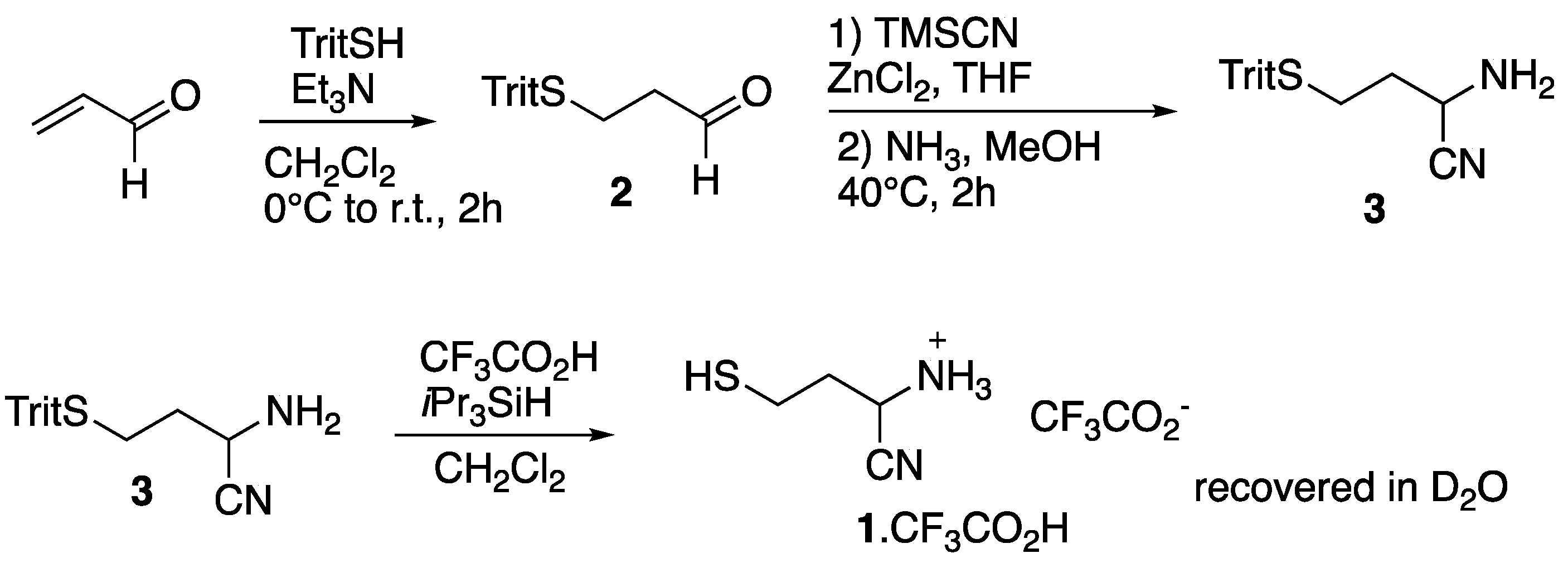{kind=link}
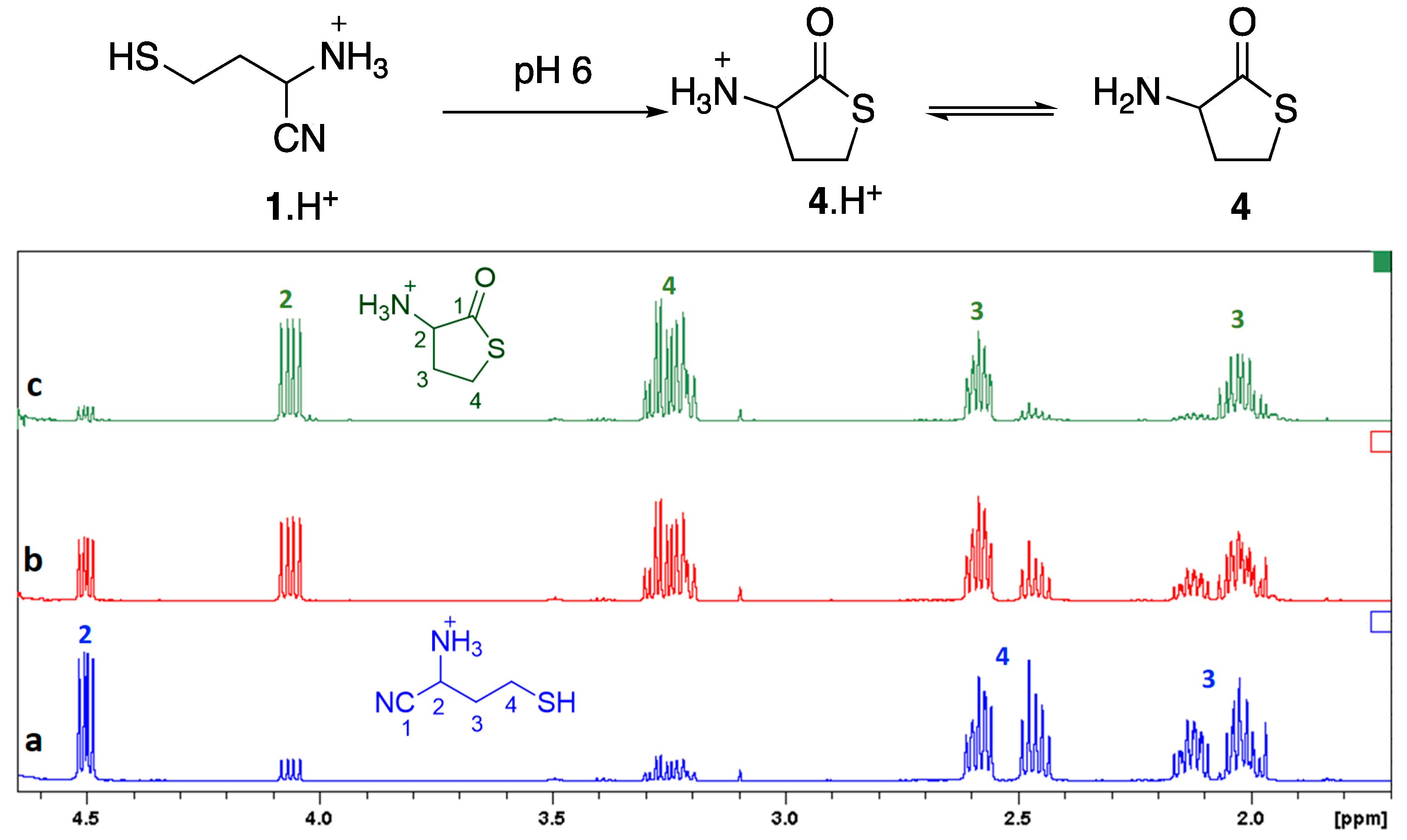{kind=link}
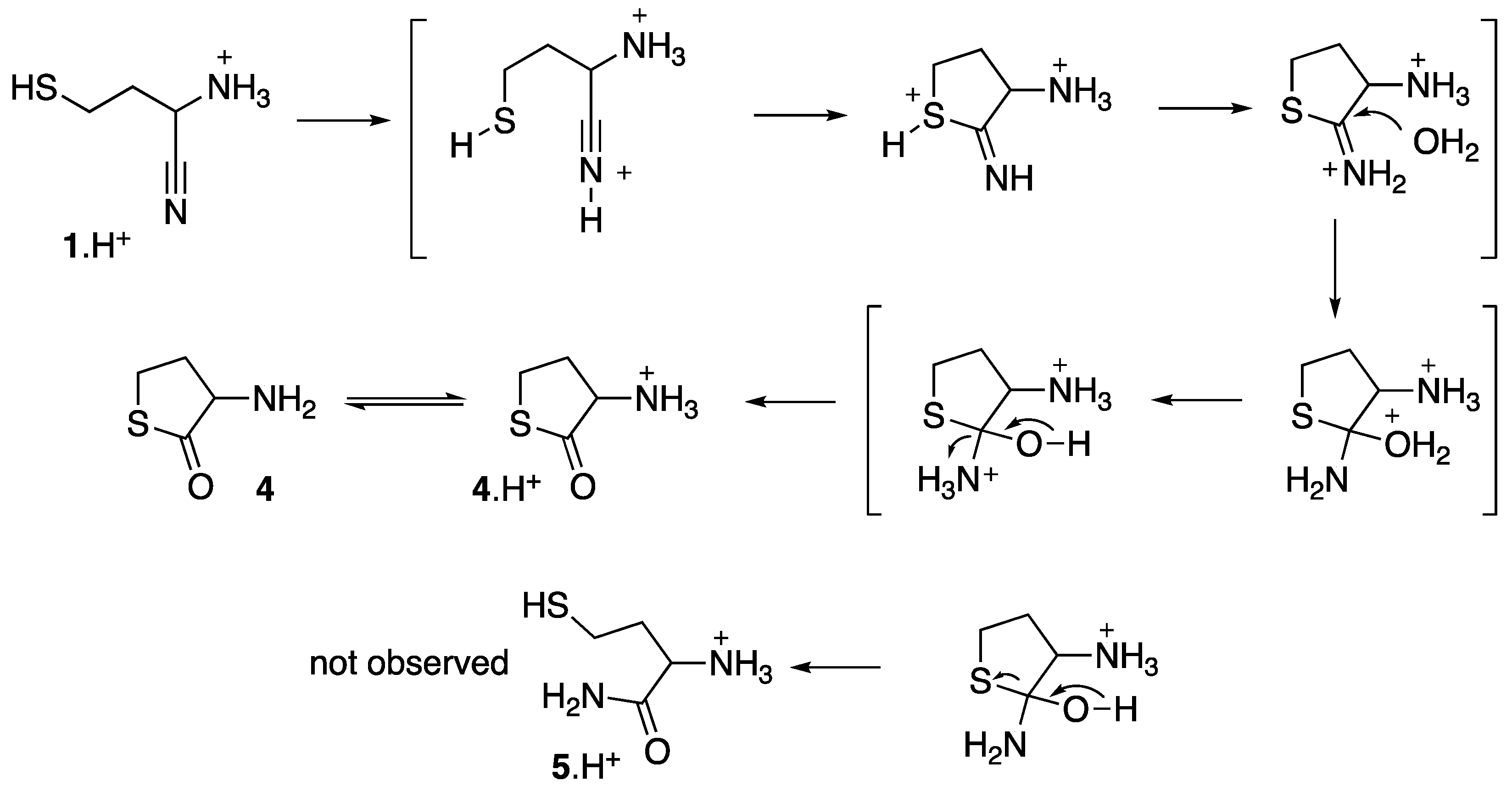{kind=link}
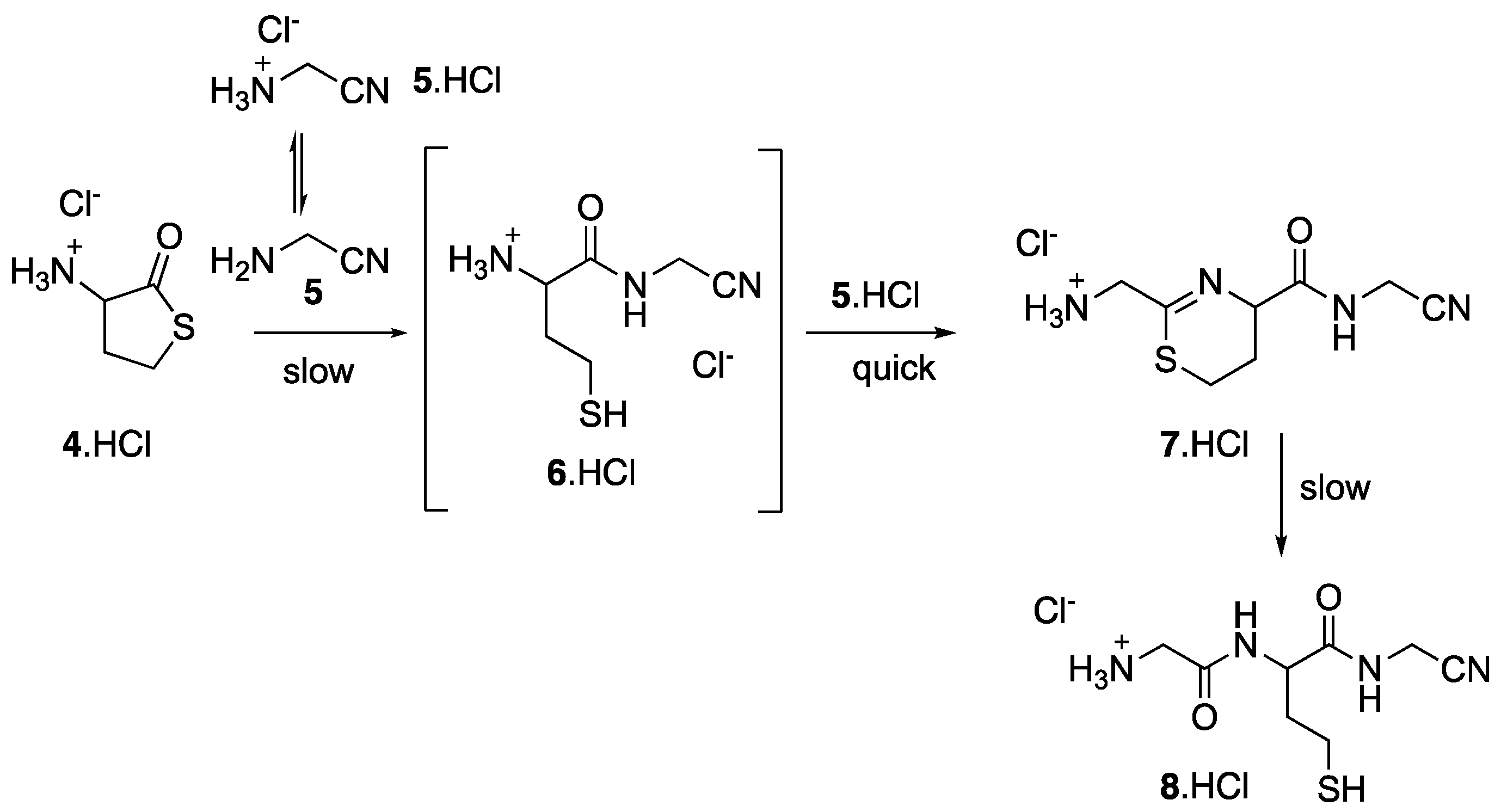{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bartlett, G.J.; Porter, C.T.; Borkakoti, N.; Thornton, J.M. Analysis of catalytic residues in enzyme active sites. J. Mol. Biol. 2002, 324, 105–121. [Google Scholar] [CrossRef]
- Buller, A.R.; Townsend, G.A. Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad. Proc. Natl. Acad. Sci. USA 2013, 110, E653–E661. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.S.; Majumdar, I.; Grishin, N.V. Structural classification of zinc fingers. Nucleic Acids Res. 2003, 31, 532–550. [Google Scholar] [CrossRef] [PubMed]
- Rawat, S.; Stemmler, T.L. Key players and their role during mitochondrial iron-sulfur cluster biosynthesis. Chem. Eur. J. 2011, 17, 746–753. [Google Scholar] [CrossRef]
- Blattner, F.R.; Plunkett, G.; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Vallée, Y.; Milet, A.; Raghavendra Rao, K.V. Was methionine the molecular trigger of life on early Earth? Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 329–332. [Google Scholar] [CrossRef]
- Jakubowski, H. Proofreading and the evolution of a methyl donor function. J. Biol. Chem. 1993, 268, 6549–6553. [Google Scholar]
- Raghavendra Rao, K.V.; Caiveau, N.; David, R.; Shalayel, I.; Milet, A.; Vallée, Y. Theoretical study, synthesis, and reactivity of five-membered-ring acyl sulfonium cations. Eur. J. Org. Chem. 2015, 28, 6125–6129. [Google Scholar]
- Perla-Kajan, J.; Twardowski, T.; Jakubowski, H. Mechanisms of homocysteine toxicity in humans. Amino Acids 2007, 32, 561–572. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine modification in protein structure/function and in human disease. Physiol. Rev. 2019, 99, 555–604. [Google Scholar] [CrossRef]
- Shalayel, I.; Coulibaly, S.; Ly, K.D.; Milet, A.; Vallée, Y. The reaction of aminonitriles with aminothiols: A way to thiol-containing peptides and nitrogen heterocycles in the primitive earth ocean. Life 2018, 8, 47. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine editing, thioester chemistry, coenzyme A, and the origin of coded peptide synthesis. Life 2017, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. A research proposal on the origin of life. Orig. Life Evol. Biosph. 2003, 33, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.I. Ribosome-independent peptide synthesis in nature and their application to dipeptide production. J. Biol. Macromol. 2008, 8, 28–37. [Google Scholar]
- Reanney, D.C. Aminoacyl thiol esters and the origins of genetic specificity. J. Theor. Biol. 1977, 65, 555–569. [Google Scholar] [CrossRef]
- Martin, R.S.; Mather, T.A.; Pyle, D.M. Volcanic emissions and the early Earth atmosphere. Geochim. Cosmochim. Acta 2007, 71, 3673–3685. [Google Scholar] [CrossRef]
- Zahnle, K.J. Photochemistry of methane and the formation of hydrocyanic acid (HCN) in the Earth’s early atmosphere. J. Geophys. Res. 1986, 91, 2819–2834. [Google Scholar] [CrossRef]
- Strecker, A. Ueber die künstliche bildung der milchsäure und einen neuen, dem glycocoll homologen körper. Liebigs Ann. Chem. 1850, 75, 27–45. [Google Scholar] [CrossRef]
- Van Trump, J.E.; Miller, S.L. Prebiotic synthesis of methionine. Science 1972, 178, 859–860. [Google Scholar] [CrossRef]
- Carlsen, L.; Egsgaard, H.; Jørgensen, F.S.; Nicolaisen, F.M. 3-Mercaptopropanal. J. Chem. Soc. Perkin Trans. II 1984, 2, 609–613. [Google Scholar] [CrossRef]
- Fuse, S.; Okada, K.; Iijima, Y.; Munakata, A.; Machida, K.; Takahashi, T.; Takagi, M.; Shin-ya, K.; Doi, T. Total synthesis of spiruchostatin B aided by an automated synthesizer. Org. Biomol. Chem. 2011, 9, 3825–3833. [Google Scholar] [CrossRef] [PubMed]
- Mai, K.; Patil, G. Facile synthesis of α-aminonitriles. Tetrahedron Lett. 1984, 25, 4583–4586. [Google Scholar] [CrossRef]
- Roger, R.; Neilson, D.G. The chemistry of imidates. Chem. Rev. 1961, 61, 179–211. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Schmir, G.L. The hydrolysis of thioimidate esters II. Evidence for the formation of three species of the tetrahedral intermediate. J. Am. Chem. Soc. 1969, 91, 737–746. [Google Scholar] [CrossRef]
- Pinti, D.L. The origin and evolution of the oceans. In Lectures in astrobiology; Gargaud, M., Barbier, B., Martin, H., Reisse, J., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2005; Volume 8, pp. 83–112. [Google Scholar]
- Chandru, K.; Gilbert, A.; Butch, C.; Aano, M.; Cleaves II, H.J. The Abiotic Chemistry of Thiolated Acetate Derivatives and the Origin of Life. Sci. Rep. 2016. [Google Scholar] [CrossRef]
- Jakubowski, H. Metabolism of homocysteine thiolactone in human cell cultures. Possible mechanism for pathological consequences of elevated homocysteine levels. J. Biol. Chem. 1997, 272, 1935–1942. [Google Scholar]
- Laliberté, R.; Knobler, Y.; Frankel, M. Amination and acylation reactions by homocysteine thiolactone and N-benzyloxycarbonylhomocysteine thiolactone. J. Chem. Soc. 1963, 2756–2762. [Google Scholar] [CrossRef]
- Song, B.J.; Jencks, W.P. Aminolysis of benzoyl fluorides in water. J. Am. Chem. Soc. 1989, 111, 8479–8484. [Google Scholar] [CrossRef]
- Jakubowski, H. Mechanism of the condensation of homocysteine thiolactone with aldehydes. Chem. Eur. J. 2006, 12, 8039–8043. [Google Scholar] [CrossRef]
- Bada, J.L. New insights into prebiotic chemistry from Stanley Miller’s spark discharge experiments. Chem. Soc. Rev. 2013, 42, 2186–2196. [Google Scholar] [CrossRef]
- Khare, B.N.; Sagan, C. Synthesis of cystine in simulated primitive conditions. Nature 1971, 232, 577–579. [Google Scholar] [CrossRef]
- Riegel, B.; du Vigneaud, V. The isolation of homocysteine and its conversion to a thiolactone. J. Biol. Chem. 1935, 112, 149–154. [Google Scholar]
- Mommer, S.; Lamberts, K.; Keul, H.; Möller, M. A novel multifunctional coupler: The concept of coupling and proof of principle. Chem. Commun. 2013, 49, 3288–3290. [Google Scholar] [CrossRef] [PubMed]
- Shalayel, I. A Plausible Prebiotic Synthesis of Thiol-Rich Peptides: The Reaction of Aminothiols with Aminonitriles. Ph.D. Thesis, University Grenoble Alpes, Grenoble, France, 2018. Available online: http://www.theses.fr/2018GREAV055 (accessed on 15 May 2019).
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–778. [Google Scholar] [CrossRef]
- Nanda, J.; Rubinov, B.; Ivnitski, D.; Mukherjee, R.; Shtelman, E.; Motro, Y.; Miler, Y.; Wagner, N.; Cohen-Luria, R.; Ashkenasy, G. Emergence of native peptide sequences in prebiotic replication networks. Nat. Commun. 2017, 8, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, B.H.; Percivalle, C.; Ritson, D.J.; Duffy, C.D.; Sutherland, J.D. Common origins of RNA, protein and lipid precursors in a cyanosulfidic protometabolism. Nat. Chem. 2015, 7, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Muchowska, K.B.; Varma, S.J.; Chevalet-Beroux, E.; Lethuillier-Karl, L.; Li, G.; Moran, J. Metals promote sequences of the reverse Krebs cycle. Nature Ecol. Evol. 2017, 1, 1716–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitadai, N.; Kameya, M.; Fujishima, K. Origin of the Reductive Tricarboxylic Acid (rTCA) Cycle-Type CO2 Fixation: A Perspective. Life 2017, 7, 39. [Google Scholar] [CrossRef]
- Bonfio, C.; Mansy, S. The chemical roots of Iron-Sulfur dependent metabolism. Biochemistry 2017, 56, 5225–5226. [Google Scholar] [CrossRef] [PubMed]
- Bracher, P.J.; Snyder, P.W.; Bohall, B.R.; Whitesides, G.M. The relative rates of thiol-thioester exchange and hydrolysis for alkyl and aryl thioalkanoates in water. Orig. Life Evol. Biosph. 2011, 41, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Semenov, S.N.; Kraft, L.J.; Ainla, A.; Zhao, M.; Baghbanzadeh, M.; Campbell, V.E.; Kang, K.; Fox, J.M.; Whitesides, G.M. Autocatalytic, bistable, oscillatory networks of biologically relevant organic reactions. Nature 2016, 537, 656–660. [Google Scholar] [CrossRef] [Green Version]













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shalayel, I.; Vallée, Y. Chemistry of Homocysteine Thiolactone in A Prebiotic Perspective. Life 2019, 9, 40. https://doi.org/10.3390/life9020040
Shalayel I, Vallée Y. Chemistry of Homocysteine Thiolactone in A Prebiotic Perspective. Life. 2019; 9(2):40. https://doi.org/10.3390/life9020040
Chicago/Turabian StyleShalayel, Ibrahim, and Yannick Vallée. 2019. "Chemistry of Homocysteine Thiolactone in A Prebiotic Perspective" Life 9, no. 2: 40. https://doi.org/10.3390/life9020040
APA StyleShalayel, I., & Vallée, Y. (2019). Chemistry of Homocysteine Thiolactone in A Prebiotic Perspective. Life, 9(2), 40. https://doi.org/10.3390/life9020040