First Glance of Molecular Profile of Atypical Cellular Angiofibroma/Cellular Angiofibroma with Sarcomatous Transformation by Next Generation Sequencing


 ,
,
Abstract
:1. Introduction
2. Method and Materials
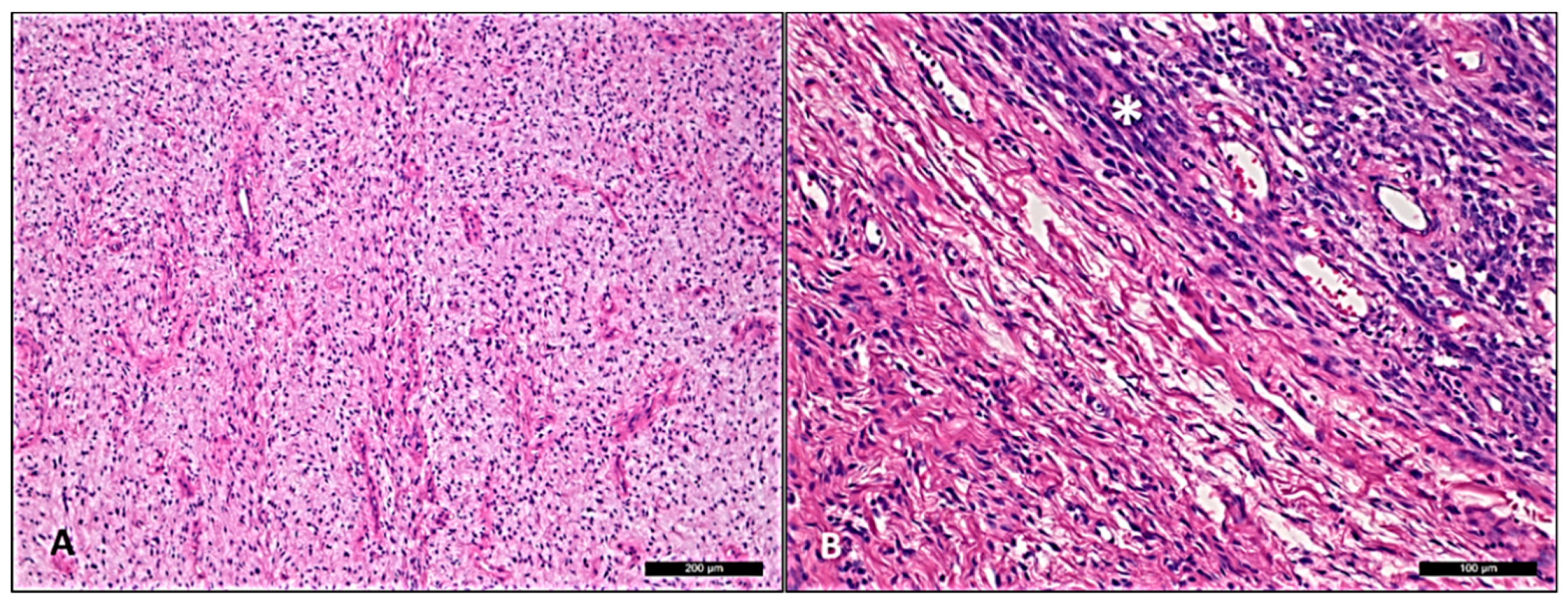
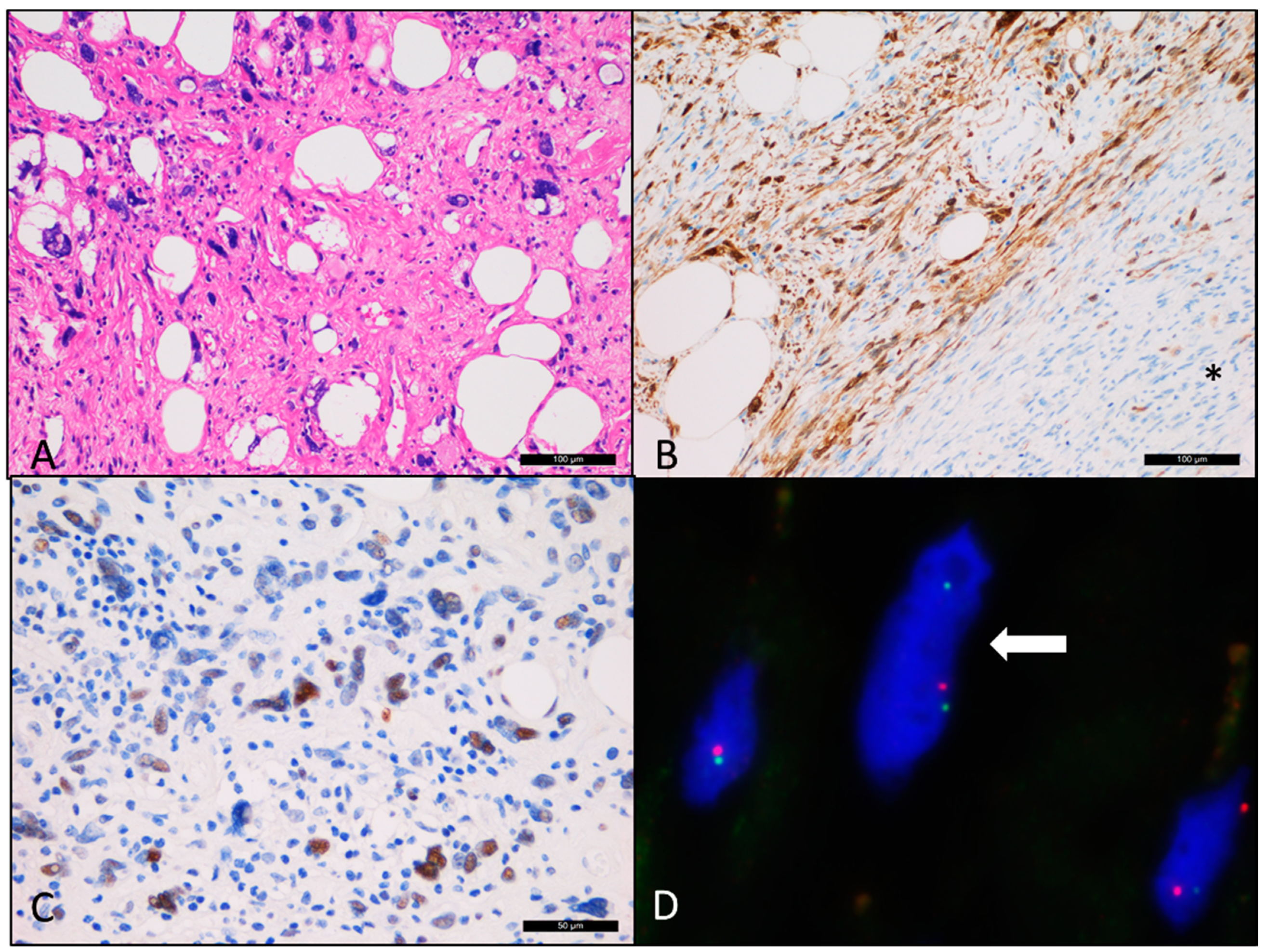
3. Result
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mandato, V.D.; Santagni, S.; Cavazza, A.; Aguzzoli, L.; Abrate, M.; La Sala, G.B. Cellular angiofibroma in women: A review of the literature. Diagn. Pathol. 2015, 10, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nucci, M.R.; Granter, S.R.; Fletcher, C.D. Cellular angiofibroma: A benign neoplasm distinct from angiomyofibroblastoma and spindle cell lipoma. Am. J. Surg. Pathol. 1997, 21, 636–644. [Google Scholar] [CrossRef]
- Uehara, K.; Ikehara, F.; Shibuya, R.; Nakazato, I.; Oshiro, M.; Kiyuna, M.; Tanabe, Y.; Toyoda, Z.; Kurima, K.; Kina, S.; et al. Molecular Signature of Tumors with Monoallelic 13q14 Deletion: A Case Series of Spindle Cell Lipoma and Genetically-Related Tumors Demonstrating a Link Between FOXO1 Status and p38 MAPK Pathway. Pathol. Oncol. Res. 2018, 24, 861–869. [Google Scholar] [CrossRef] [Green Version]
- Hameed, M.; Clarke, K.; Amer, H.Z.; Mahmet, K.; Aisner, S. Cellular angiofibroma is genetically similar to spindle cell lipoma: A case report. Cancer Genet. Cytogenet. 2007, 177, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Creytens, D.; van Gorp, J.; Savola, S.; Ferdinande, L.; Mentzel, T.; Libbrecht, L. Atypical spindle cell lipoma: A clinicopathologic, immunohistochemical, and molecular study emphasizing its relationship to classical spindle cell lipoma. Virchows Arch. 2014, 465, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Fletcher, C.D. Cellular angiofibroma with atypia or sarcomatous transformation: Clinicopathologic analysis of 13 cases. Am. J. Surg. Pathol. 2010, 34, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.; Serrano, M. A new mechanism of inactivation of the INK4/ARF locus. Cell Cycle 2006, 5, 1382–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, E.; Saeb, M.; Crum, C.P.; Woo, S.B.; McKee, P.H.; Rheinwald, J.G. Co-expression of p16(INK4A) and laminin 5 gamma2 by microinvasive and superficial squamous cell carcinomas in vivo and by migrating wound and senescent keratinocytes in culture. Am. J. Pathol. 2003, 163, 477–491. [Google Scholar] [CrossRef]
- Reuschenbach, M.; Waterboer, T.; Wallin, K.L.; Einenkel, J.; Dillner, J.; Hamsikova, E.; Eschenbach, D.; Zimmer, H.; Heilig, B.; Kopitz, J.; et al. Characterization of humoral immune responses against p16, p53, HPV16 E6 and HPV16 E7 in patients with HPV-associated cancers. Int. J. Cancer 2008, 123, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Horrée, N.; van Diest, P.J.; Sie-Go, D.M.; Heintz, A.P. The invasive front in endometrial carcinoma: Higher proliferation and associated derailment of cell cycle regulators. Hum. Pathol. 2007, 38, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Val-Bernal, J.F.; Azueta, A.; Parra, A.; Mediavilla, E.; Zubillaga, S. Paratesticular cellular angiofibroma with atypical (bizarre) cells: Case report and literature review. Pathol. Res. Pract. 2013, 209, 388–392. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Case No. | Gender | Age | Location | Size (cm) | Depth | Atypical Areas | 50/HPF | Atypical MF | Necrosis (%) | Infiltration | ER/PR | CD34 | p16 | p53 | RB1 FISH | Follow-up |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 68 | pelvis | 4 | deep | CAS with PLPS-like areas | 2 | no | 0 | circumscribed | 1+ | 3+ | 3+ | negative | positive | 16 months |
| 2 | M | 36 | hip | 9 | deep | CAS with UPS-like areas | 8 | no | 0 | mixed | 1+ | 3+ | 3+ | positive | positive | 12 months |
| 3 | M | 73 | inguinal | 3.5 | superficial | CAS with PLPS-like areas | 0 | no | 0 | circumscribed | 3+ | 3+ | 2+ | negative | positive | 10 months |
| 4 | F | 48 | perineum | 2.3 | superficial | CAS with UPS-like areas | 4 | no | 0 | circumscribed | 3+ | 3+ | 2+ | negative | positive | 12 months |
| 5 | F | 25 | vulva | 4.3 | deep | ACA | 0 | no | 0 | circumscribed | 3+ | 1+ | 2+ | negative | positive | 8 months |
| 6 | F | 43 | vulva | 7 | deep | CAS with PLPS-like areas | 6 | no | 0 | circumscribed | 3+ | 3+ | 2+ | positive | positive | 6 months |
| CA1 | M | 63 | inguinal | 7.5 | deep | 2 | no | 0 | circumscribed | 1+ | 2+ | negative | negative | positive | 12 months | |
| CA2 | F | 52 | vulva | 2.2 | superficial | 0 | no | 0 | circumscribed | 1+ | 2+ | negative | negative | positive | 24 months |
| Case No. | Gene | Mutation | Transcript IDs | VAF (%) | Significance | gnomAD Frequency (%) |
|---|---|---|---|---|---|---|
| 1 | SMO | c.743C>T; p.Thr248Ile | NM_005631.4 | 2.5 | Uncertain | - |
| RHOA | c.178A>G; p.Thr60Ala | NM_001313941.1 | 2.3 | Uncertain | - | |
| 2 | PTEN | c.511C>G; p.Leu171Val | NM_001304717.5 | 53 | Heterozygous, benign | 0.4 |
| TP53 | c.626_627del; p.Arg209LysfsTer6 | NM_000546.5 | 39.2 | Pathogenic | - | |
| H3F3A | c.89C>T; p.Ala30Val | NM_002107.4 | 3.4 | Uncertain | - | |
| PIK3CA | c.916T>C; p.Ser306Pro | NM_006218.2 | 3 | Uncertain | - | |
| 3 | CDH1 | c.1174C>T; p.His392Tyr | NM_001317184.1 | 3.1 | Uncertain | - |
| TP53 | c.3G>A; p.Met1 | NM_001126118.1 | 2.8 | Uncertain | - | |
| APC | c.6710G>A; p.Arg2237Gln | NM_000038.5 | 2.4 | Uncertain | - | |
| KIT | c.148G>A; p.Val50Met | NM_000222.2 | 2 | Benign | 0.001 | |
| 4 | FBXW7 | c.732T>G; p.Asp244Glu | NM_001013415.1 | 4.4 | Uncertain | - |
| PIK3CA | c.44T>G; p.Leu15Trp | NM_006218.2 | 3.4 | Uncertain | - | |
| HNF1A | c.872dup; p.Gly292ArgfsTer25 | NM_000545.7 | 2.1 | Pathogenic | 0.03 | |
| 5 | STK11 | c.1062C>G; p.Phe354Leu | NM_000455.5 | 56 | Heterozygous, benign | 0.5 |
| ALK | c.3385G>A; p.Glu1129Lys | NM_004304.4 | 3 | Pathogenic | - | |
| EGFR | c.2340G>A; p.Met780Ile | NM_001346897.1 | 2.7 | Uncertain | - | |
| MET | c.3943G>A; p.Val1315Ile | NM_000245.2 | 2.4 | Uncertain | - | |
| TP53 | c.328C>T; p.Arg110Cys | NM_000546.5 | 2 | Benign | 0.002 | |
| 6 | TP53 | c.488A>G; Tyr163Cys | NM_000546.5 | 63.6 | Pathogenic | - |
| SMO | c.1097C>T; p.Ser366Leu | NM_005631.4 | 4.6 | Uncertain | - | |
| JAK3 | c.2141C>T; p.Thr714Met | NM_000215.3 | 4.5 | Uncertain | 0.004 | |
| MYC | c.1240G>A; p.Ala414Thr | NM_002467.4 | 3 | Uncertain | - | |
| NRAS | c.436G>A; p.Ala146Thr | NM_002524.4 | 2.8 | Pathogenic | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang Chien, Y.-C.; Mokánszki, A.; Huang, H.-Y.; Silva, R.G., Jr.; Chen, C.-C.; Beke, L.; Mónus, A.; Méhes, G. First Glance of Molecular Profile of Atypical Cellular Angiofibroma/Cellular Angiofibroma with Sarcomatous Transformation by Next Generation Sequencing. Diagnostics 2020, 10, 35. https://doi.org/10.3390/diagnostics10010035
Chang Chien Y-C, Mokánszki A, Huang H-Y, Silva RG Jr., Chen C-C, Beke L, Mónus A, Méhes G. First Glance of Molecular Profile of Atypical Cellular Angiofibroma/Cellular Angiofibroma with Sarcomatous Transformation by Next Generation Sequencing. Diagnostics. 2020; 10(1):35. https://doi.org/10.3390/diagnostics10010035
Chicago/Turabian StyleChang Chien, Yi-Che, Attila Mokánszki, Hsuan-Ying Huang, Raimundo Geronimo Silva, Jr., Chien-Chin Chen, Lívia Beke, Anikó Mónus, and Gábor Méhes. 2020. "First Glance of Molecular Profile of Atypical Cellular Angiofibroma/Cellular Angiofibroma with Sarcomatous Transformation by Next Generation Sequencing" Diagnostics 10, no. 1: 35. https://doi.org/10.3390/diagnostics10010035
APA StyleChang Chien, Y. -C., Mokánszki, A., Huang, H. -Y., Silva, R. G., Jr., Chen, C. -C., Beke, L., Mónus, A., & Méhes, G. (2020). First Glance of Molecular Profile of Atypical Cellular Angiofibroma/Cellular Angiofibroma with Sarcomatous Transformation by Next Generation Sequencing. Diagnostics, 10(1), 35. https://doi.org/10.3390/diagnostics10010035