The Multifaceted Roles of MicroRNAs in Cystic Fibrosis

 ,
,  and
and
Abstract
:1. Introduction
2. Cystic Fibrosis
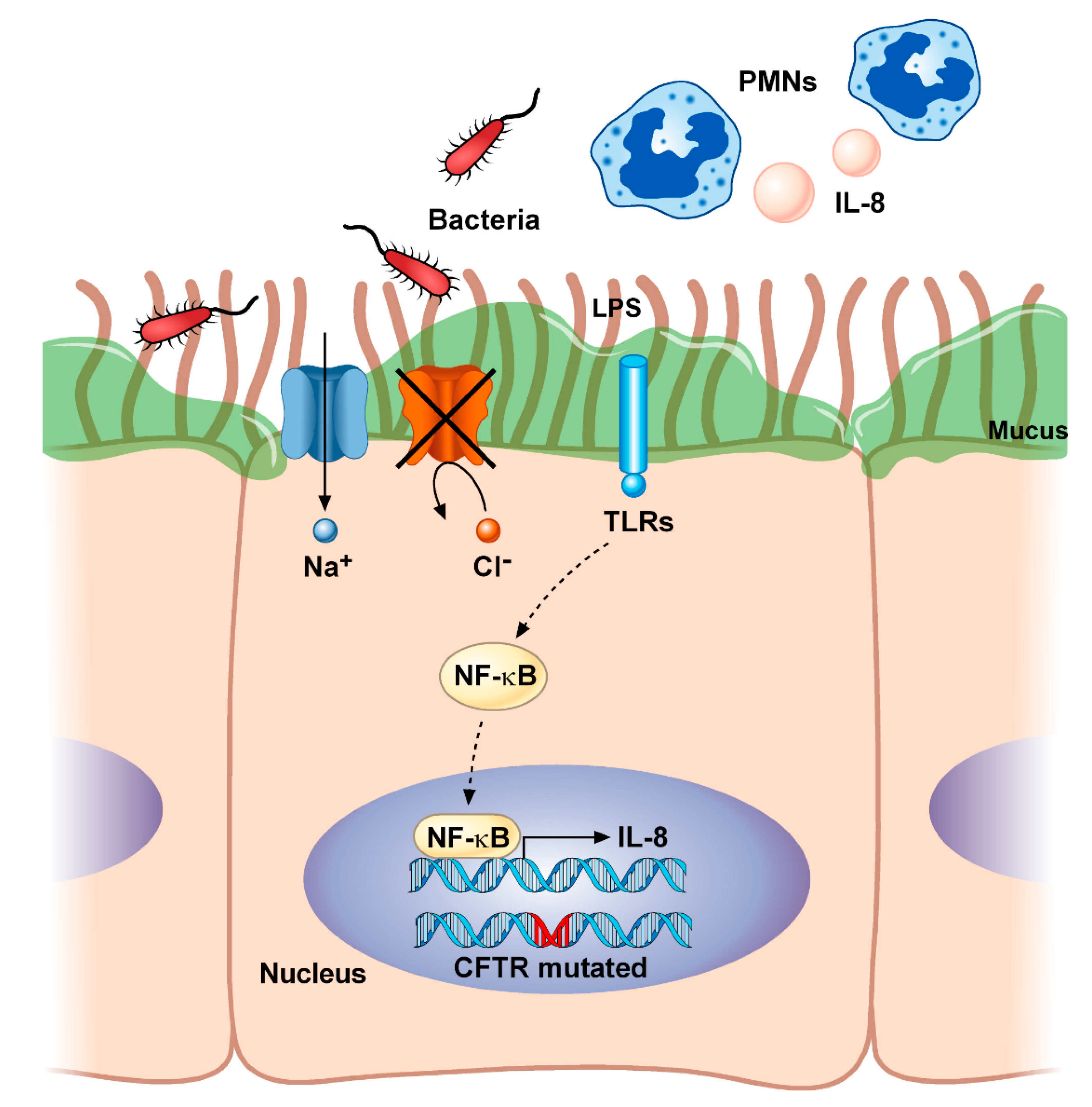
2.1. Cystic Fibrosis, A Multisystemic Deadly Disease
2.2. The Mutational Landscape of the Cystic Fibrosis
3. microRNAs in Cystic Fibrosis
Altered microRNAs in the Regulation of CFTR
4. microRNAs as Regulators of Inflammation in Cystic Fibrosis
5. Circulating microRNAs as Potential Biomarkers in Cystic Fibrosis
6. Single Nucleotide Polymorphisms (SNPs) in microRNAs Targeting the CFTR Gene
7. Therapeutic Modulation of microRNAs in Cystic Fibrosis
7.1. microRNA Therapeutics
7.2. microRNA Therapeutics in Cystic Fibrosis
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cech, T.R.; Steitz, J.A. The Noncoding RNA Revolution—Trashing Old Rules to Forge New Ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Witmer, P.D.; Casey, E.; Valle, D.; Sukumar, S. DNA methylation regulates MicroRNA expression. Cancer Biol. 2007, 6, 1284–1288. [Google Scholar] [CrossRef]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef] [Green Version]
- Lan, H.; Lu, H.; Wang, X.; Jin, H. MicroRNAs as Potential Biomarkers in Cancer: Opportunities and Challenges. Available online: https://www.hindawi.com/journals/bmri/2015/125094/ (accessed on 3 September 2020).
- De Palma, F.D.E.; Luglio, G.; Tropeano, F.P.; Pagano, G.; D’Armiento, M.; Kroemer, G.; Maiuri, M.C.; De Palma, G.D. The Role of Micro-RNAs and Circulating Tumor Markers as Predictors of Response to Neoadjuvant Therapy in Locally Advanced Rectal Cancer. Int. J. Mol. Sci. 2020, 21, 7040. [Google Scholar] [CrossRef]
- De Palma, F.D.E.; D’Argenio, V.; Pol, J.; Kroemer, G.; Maiuri, M.C.; Salvatore, F. The Molecular Hallmarks of the Serrated Pathway in Colorectal Cancer. Cancers 2019, 11, 1017. [Google Scholar] [CrossRef] [Green Version]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Li, Z.; Rana, T.M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef] [PubMed]
- Finotti, A.; Fabbri, E.; Lampronti, I.; Gasparello, J.; Borgatti, M.; Gambari, R. MicroRNAs and Long Non-coding RNAs in Genetic Diseases. Mol. Diagn. 2019, 23, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Narożna, B.; Langwiński, W.; Szczepankiewicz, A. Non-Coding RNAs in Pediatric Airway Diseases. Genes 2017, 8, 348. [Google Scholar] [CrossRef] [Green Version]
- Greene, C.M. MicroRNA Expression in Cystic Fibrosis Airway Epithelium. Biomolecules 2013, 3, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varilh, J.; Bonini, J.; Taulan-Cadars, M. Role of Non-coding RNAs in Cystic Fibrosis. Cyst. Fibros. Light New Res. 2015. [Google Scholar] [CrossRef] [Green Version]
- Sonneville, F.; Ruffin, M.; Guillot, L.; Rousselet, N.; Le Rouzic, P.; Corvol, H.; Tabary, O. New insights about miRNAs in cystic fibrosis. Am. J. Pathol. 2015, 185, 897–908. [Google Scholar] [CrossRef]
- Noel, S.; Leal, T. Emerging Roles of microRNAs in Cystic Fibrosis—From Pathogenesis to Development of New Therapies. Cyst. Fibros. Light New Res. 2015. [Google Scholar] [CrossRef] [Green Version]
- Mitash, N.; Donovan, J.E.; Swiatecka-Urban, A. The Role of MicroRNA in the Airway Surface Liquid Homeostasis. Int. J. Mol. Sci. 2020, 21, 3848. [Google Scholar] [CrossRef]
- McKiernan, P.J.; Greene, C.M. MicroRNA Dysregulation in Cystic Fibrosis. Available online: https://www.hindawi.com/journals/mi/2015/529642/ (accessed on 25 November 2020).
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Scotet, V.; L’Hostis, C.; Férec, C. The Changing Epidemiology of Cystic Fibrosis: Incidence, Survival and Impact of the CFTR Gene Discovery. Genes 2020, 11, 589. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.-C.; Kirk, K.L. The CFTR Ion Channel: Gating, Regulation, and Anion Permeation. Cold Spring Harb. Perspect. Med. 2013, 3. [Google Scholar] [CrossRef] [Green Version]
- Ratjen, F.; Bell, S.C.; Rowe, S.M.; Goss, C.H.; Quittner, A.L.; Bush, A. Cystic fibrosis. Nat. Rev. Dis. Primers 2015, 1, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell. Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marson, F.A.L.; Bertuzzo, C.S.; Ribeiro, J.D. Classification of CFTR mutation classes. Lancet Respir. Med. 2016, 4, e37–e38. [Google Scholar] [CrossRef] [Green Version]
- Puchelle, E.; Bajolet, O.; Abély, M. Airway mucus in cystic fibrosis. Paediatr. Respir. Rev. 2002, 3, 115–119. [Google Scholar] [CrossRef]
- Chioccioli, M.; Feriani, L.; Kotar, J.; Bratcher, P.E.; Cicuta, P. Phenotyping ciliary dynamics and coordination in response to CFTR-modulators in Cystic Fibrosis respiratory epithelial cells. Nat. Commun. 2019, 10, 1763. [Google Scholar] [CrossRef] [Green Version]
- Françoise, A.; Héry-Arnaud, G. The Microbiome in Cystic Fibrosis Pulmonary Disease. Genes 2020, 11, 536. [Google Scholar] [CrossRef]
- Coburn, B.; Wang, P.W.; Diaz Caballero, J.; Clark, S.T.; Brahma, V.; Donaldson, S.; Zhang, Y.; Surendra, A.; Gong, Y.; Elizabeth Tullis, D.; et al. Lung microbiota across age and disease stage in cystic fibrosis. Sci. Rep. 2015, 5, 10241. [Google Scholar] [CrossRef]
- Ooi, C.Y.; Durie, P.R. Cystic fibrosis from the gastroenterologist’s perspective. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 175–185. [Google Scholar] [CrossRef]
- De Lisle, R.C.; Borowitz, D. The cystic fibrosis intestine. Cold Spring Harb. Perspect. Med. 2013, 3, a009753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagolan, P.; Morini, F.; Conforti, A. Meconium Ileus. In Pediatric Surgery: General Principles and Newborn Surgery; Puri, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2020; pp. 973–992. ISBN 978-3-662-43588-5. [Google Scholar]
- Hayden, H.S.; Eng, A.; Pope, C.E.; Brittnacher, M.J.; Vo, A.T.; Weiss, E.J.; Hager, K.R.; Martin, B.D.; Leung, D.H.; Heltshe, S.L.; et al. Fecal dysbiosis in infants with cystic fibrosis is associated with early linear growth failure. Nat. Med. 2020, 26, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Madácsy, T.; Pallagi, P.; Maleth, J. Cystic Fibrosis of the Pancreas: The Role of CFTR Channel in the Regulation of Intracellular Ca2+ Signaling and Mitochondrial Function in the Exocrine Pancreas. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casciaro, R.; Cresta, F.; Favilli, F.; Minicucci, L. Cystic Fibrosis and Fertility. Cyst. Fibros. Light New Res. 2015. [Google Scholar] [CrossRef] [Green Version]
- Kayani, K.; Mohammed, R.; Mohiaddin, H. Cystic Fibrosis-Related Diabetes. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C. Cystic fibrosis-related diabetes | Nature Reviews Endocrinology. Nat. Rev. Endocrinol. 2011, 7, 375. [Google Scholar] [CrossRef]
- Bieth, E.; Hamdi, S.M.; Mieusset, R. Genetics of the congenital absence of the vas deferens. Hum. Genet. 2020. [Google Scholar] [CrossRef] [Green Version]
- de Souza, D.A.S.; Faucz, F.R.; Pereira-Ferrari, L.; Sotomaior, V.S.; Raskin, S. Congenital Bilateral Absence of the Vas Deferens as an Atypical Form of Cystic Fibrosis: Reproductive Implications and Genetic Counseling. Andrology 2018, 6, 127–135. [Google Scholar] [CrossRef]
- Wagenknecht, L.V.; Lotzin, C.F.; Sommer, H.-J.; Schirren, C. Vas Deferens Aplasia: Clinical and Anatomical Features of 90 Cases. Andrologia 1983, 15, 605–613. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharm. 2020, 10. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, L.; Raia, V.; Kroemer, G. Strategies for the etiological therapy of cystic fibrosis. Cell Death Differ. 2017, 24, 1825–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeck, K.D.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- Ramos, K.J.; Smith, P.J.; McKone, E.F.; Pilewski, J.M.; Lucy, A.; Hempstead, S.E.; Tallarico, E.; Faro, A.; Rosenbluth, D.B.; Gray, A.L.; et al. Lung transplant referral for individuals with cystic fibrosis: Cystic Fibrosis Foundation consensus guidelines. J. Cyst. Fibros. 2019, 18, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Fisman, D. Cystic fibrosis heterozygosity: Carrier state or haploinsufficiency? Proc. Natl. Acad. Sci. USA 2020, 117, 2740–2742. [Google Scholar] [CrossRef] [PubMed]
- Guimbellot, J.S.; Erickson, S.W.; Mehta, T.; Wen, H.; Page, G.P.; Sorscher, E.J.; Hong, J.S. Correlation of microRNA levels during hypoxia with predicted target mRNAs through genome-wide microarray analysis. BMC Med. Genom. 2009, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Bartoszewska, S.; Kamysz, W.; Jakiela, B.; Sanak, M.; Króliczewski, J.; Bebok, Z.; Bartoszewski, R.; Collawn, J.F. miR-200b downregulates CFTR during hypoxia in human lung epithelial cells. Cell. Mol. Biol. Lett. 2017, 22, 23. [Google Scholar] [CrossRef] [Green Version]
- Megiorni, F.; Cialfi, S.; Dominici, C.; Quattrucci, S.; Pizzuti, A. Synergistic post-transcriptional regulation of the Cystic Fibrosis Transmembrane conductance Regulator (CFTR) by miR-101 and miR-494 specific binding. PLoS ONE 2011, 6, e26601. [Google Scholar] [CrossRef]
- Hassan, F.; Nuovo, G.J.; Crawford, M.; Boyaka, P.N.; Kirkby, S.; Nana-Sinkam, S.P.; Cormet-Boyaka, E. MiR-101 and miR-144 Regulate the Expression of the CFTR Chloride Channel in the Lung. PLoS ONE 2012, 7, e50837. [Google Scholar] [CrossRef] [Green Version]
- Viart, V.; Bergougnoux, A.; Bonini, J.; Varilh, J.; Chiron, R.; Tabary, O.; Molinari, N.; Claustres, M.; Taulan-Cadars, M. Transcription factors and miRNAs that regulate fetal to adult CFTR expression change are new targets for cystic fibrosis. Eur. Respir. J. 2015, 45, 116–128. [Google Scholar] [CrossRef] [Green Version]
- Gillen, A.E.; Gosalia, N.; Leir, S.-H.; Harris, A. MicroRNA regulation of expression of the cystic fibrosis transmembrane conductance regulator gene. Biochem. J. 2011, 438, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Karp, P.H.; Osterhaus, S.R.; Jiang, P.; Wohlford-Lenane, C.; Lennox, K.A.; Jacobi, A.M.; Praekh, K.; Rose, S.D.; Behlke, M.A.; et al. Post-transcriptional regulation of cystic fibrosis transmembrane conductance regulator expression and function by microRNAs. Am. J. Respir. Cell Mol. Biol. 2013, 49, 544–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, F.; Seia, M.; Giordano, S.; Elce, A.; Zarrilli, F.; Castaldo, G.; Tomaiuolo, R. Gene Mutation in MicroRNA Target Sites of CFTR Gene: A Novel Pathogenetic Mechanism in Cystic Fibrosis? PLoS ONE 2013, 8, e60448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonneville, F.; Ruffin, M.; Coraux, C.; Rousselet, N.; Le Rouzic, P.; Blouquit-Laye, S.; Corvol, H.; Tabary, O. MicroRNA-9 downregulates the ANO1 chloride channel and contributes to cystic fibrosis lung pathology. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Ramachandran, S.; Karp, P.H.; Jiang, P.; Ostedgaard, L.S.; Walz, A.E.; Fisher, J.T.; Keshavjee, S.; Lennox, K.A.; Jacobi, A.M.; Rose, S.D.; et al. A microRNA network regulates expression and biosynthesis of wild-type and ΔF508 mutant cystic fibrosis transmembrane conductance regulator. PNAS 2012, 109, 13362–13367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedetto, R.; Ousingsawat, J.; Wanitchakool, P.; Zhang, Y.; Holtzman, M.J.; Amaral, M.; Rock, J.R.; Schreiber, R.; Kunzelmann, K. Epithelial Chloride Transport by CFTR Requires TMEM16A. Sci. Rep. 2017, 7, 12397. [Google Scholar] [CrossRef] [Green Version]
- Oglesby, I.K.; Chotirmall, S.H.; McElvaney, N.G.; Greene, C.M. Regulation of cystic fibrosis transmembrane conductance regulator by microRNA-145, -223, and -494 is altered in ΔF508 cystic fibrosis airway epithelium. J. Immunol. 2013, 190, 3354–3362. [Google Scholar] [CrossRef]
- Lutful Kabir, F.; Ambalavanan, N.; Liu, G.; Li, P.; Solomon, G.M.; Lal, C.V.; Mazur, M.; Halloran, B.; Szul, T.; Gerthoffer, W.T.; et al. MicroRNA-145 Antagonism Reverses TGF-β Inhibition of F508del CFTR Correction in Airway Epithelia. Am. J. Respir. Crit. Care Med. 2018, 197, 632–643. [Google Scholar] [CrossRef]
- Mitash, N.; Mu, F.; Donovan, J.E.; Myerburg, M.M.; Ranganathan, S.; Greene, C.M.; Swiatecka-Urban, A. Transforming Growth Factor-β1 Selectively Recruits microRNAs to the RNA-Induced Silencing Complex and Degrades CFTR mRNA under Permissive Conditions in Human Bronchial Epithelial Cells. Int. J. Mol. Sci. 2019, 20, 4933. [Google Scholar] [CrossRef] [Green Version]
- Fenker, D.E.; McDaniel, C.T.; Panmanee, W.; Panos, R.J.; Sorscher, E.J.; Sabusap, C.; Clancy, J.P.; Hassett, D.J. A Comparison between Two Pathophysiologically Different yet Microbiologically Similar Lung Diseases: Cystic Fibrosis and Chronic Obstructive Pulmonary Disease. Int. J. Respir. Pulm. Med. 2018, 5. [Google Scholar] [CrossRef] [Green Version]
- Dutta, R.K.; Chinnapaiyan, S.; Rasmussen, L.; Raju, S.V.; Unwalla, H.J. A Neutralizing Aptamer to TGFBR2 and miR-145 Antagonism Rescue Cigarette Smoke- and TGF-β-Mediated CFTR Expression. Mol. Ther. 2019, 27, 442–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villella, V.R.; Esposito, S.; Ferrari, E.; Monzani, R.; Tosco, A.; Rossin, F.; Castaldo, A.; Silano, M.; Marseglia, G.L.; Romani, L.; et al. Autophagy suppresses the pathogenic immune response to dietary antigens in cystic fibrosis. Cell Death Dis. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, M.C.; Kroemer, G. Therapeutic modulation of autophagy: Which disease comes first? Cell Death Differ. 2019, 26, 680–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tazi, M.F.; Dakhlallah, D.A.; Caution, K.; Gerber, M.M.; Chang, S.-W.; Khalil, H.; Kopp, B.T.; Ahmed, A.E.; Krause, K.; Davis, I.; et al. Elevated Mirc1/Mir17-92 cluster expression negatively regulates autophagy and CFTR (cystic fibrosis transmembrane conductance regulator) function in CF macrophages. Autophagy 2016, 12, 2026–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in cystic fibrosis: An update. Pediatr. Pulmonol. 2018, 53, S30–S50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karp, C.L.; Flick, L.M.; Park, K.W.; Softic, S.; Greer, T.M.; Keledjian, R.; Yang, R.; Uddin, J.; Guggino, W.B.; Atabani, S.F.; et al. Defective lipoxin-mediated anti-inflammatory activity in the cystic fibrosis airway. Nat. Immunol. 2004, 5, 388–392. [Google Scholar] [CrossRef]
- Pierdomenico, A.M.; Patruno, S.; Codagnone, M.; Simiele, F.; Mari, V.C.; Plebani, R.; Recchiuti, A.; Romano, M. microRNA-181b is increased in cystic fibrosis cells and impairs lipoxin A 4 receptor-dependent mechanisms of inflammation resolution and antimicrobial defense. Sci. Rep. 2017, 7, 13519. [Google Scholar] [CrossRef]
- Luly, F.R.; Lévêque, M.; Licursi, V.; Cimino, G.; Martin-Chouly, C.; Théret, N.; Negri, R.; Cavinato, L.; Ascenzioni, F.; Del Porto, P. MiR-146a is over-expressed and controls IL-6 production in cystic fibrosis macrophages. Sci. Rep. 2019, 9, 16259. [Google Scholar] [CrossRef] [Green Version]
- Jundi, K.; Greene, C.M. Transcription of Interleukin-8: How Altered Regulation Can Affect Cystic Fibrosis Lung Disease. Biomolecules 2015, 5, 1386–1398. [Google Scholar] [CrossRef]
- Guan, X.; Hou, Y.; Sun, F.; Yang, Z.; Li, C. Dysregulated Chemokine Signaling in Cystic Fibrosis Lung Disease: A Potential Therapeutic Target. Curr. Drug. Targets 2016, 17, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Oglesby, I.K.; Vencken, S.F.; Agrawal, R.; Gaughan, K.; Molloy, K.; Higgins, G.; McNally, P.; McElvaney, N.G.; Mall, M.A.; Greene, C.M. miR-17 overexpression in cystic fibrosis airway epithelial cells decreases interleukin-8 production. Eur. Respir. J. 2015, 46, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, E.; Borgatti, M.; Montagner, G.; Bianchi, N.; Finotti, A.; Lampronti, I.; Bezzerri, V.; Dechecchi, M.C.; Cabrini, G.; Gambari, R. Expression of microRNA-93 and Interleukin-8 during Pseudomonas aeruginosa–Mediated Induction of Proinflammatory Responses. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1144–1155. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Balakathiresan, N.S.; Dalgard, C.; Gutti, U.; Armistead, D.; Jozwik, C.; Srivastava, M.; Pollard, H.B.; Biswas, R. Elevated miR-155 promotes inflammation in cystic fibrosis by driving hyperexpression of interleukin-8. J. Biol. Chem. 2011, 286, 11604–11615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Kumar, P.; Tsuchiya, M.; Bhattacharyya, A.; Biswas, R. Regulation of miR-155 biogenesis in cystic fibrosis lung epithelial cells: Antagonistic role of two mRNA-destabilizing proteins, KSRP and TTP. Biochem. Biophys. Res. Commun. 2013, 433, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, M.; Kalurupalle, S.; Kumar, P.; Ghoshal, S.; Zhang, Y.; Lehrmann, E.; Becker, K.G.; Gorospe, M.; Biswas, R. RPTOR, a novel target of miR-155, elicits a fibrotic phenotype of cystic fibrosis lung epithelium by upregulating CTGF. RNA Biol. 2016, 13, 837–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardin, P.; Marchal-Duval, E.; Sonneville, F.; Blouquit-Laye, S.; Rousselet, N.; Rouzic, P.L.; Corvol, H.; Tabary, O. Small RNA and transcriptome sequencing reveal the role of miR-199a-3p in inflammatory processes in cystic fibrosis airways. J. Pathol. 2018, 245, 410–420. [Google Scholar] [CrossRef]
- Zhang, P.; Cheng, J.; Zou, S.; D’Souza, A.D.; Koff, J.L.; Lu, J.; Lee, P.J.; Krause, D.S.; Egan, M.E.; Bruscia, E.M. Pharmacological modulation of the AKT/microRNA-199a-5p/CAV1 pathway ameliorates cystic fibrosis lung hyper-inflammation. Nat. Commun. 2015, 6, 6221. [Google Scholar] [CrossRef] [Green Version]
- Ge, Q.; Moir, L.M.; Black, J.L.; Oliver, B.G.; Burgess, J.K. TGFβ1 induces IL-6 and inhibits IL-8 release in human bronchial epithelial cells: The role of Smad2/3. J. Cell. Physiol. 2010, 225, 846–854. [Google Scholar] [CrossRef]
- Oglesby, I.K.; Bray, I.M.; Chotirmall, S.H.; Stallings, R.L.; O’Neill, S.J.; McElvaney, N.G.; Greene, C.M. miR-126 Is Downregulated in Cystic Fibrosis Airway Epithelial Cells and Regulates TOM1 Expression. J. Immunol. 2010, 184, 1702–1709. [Google Scholar] [CrossRef] [Green Version]
- Stolzenburg, L.R.; Wachtel, S.; Dang, H.; Harris, A. miR-1343 attenuates pathways of fibrosis by targeting the TGF-β receptors. Biochem. J. 2016, 473, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Zhong, T.; Perelman, J.M.; Kolosov, V.P.; Zhou, X. MiR-146a negatively regulates neutrophil elastase-induced MUC5AC secretion from 16HBE human bronchial epithelial cells. Mol. Cell. Biochem. 2011, 358, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.M.P.; Boucher, R.C. Role of Endoplasmic Reticulum Stress in Cystic Fibrosis–Related Airway Inflammatory Responses. Proc. Am. Thorac. Soc. 2010, 7, 387–394. [Google Scholar] [CrossRef]
- Oglesby, I.K.; Agrawal, R.; Mall, M.A.; McElvaney, N.G.; Greene, C.M. miRNA-221 is elevated in cystic fibrosis airway epithelial cells and regulates expression of ATF6. Mol. Cell. Pediatr. 2015, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weldon, S.; McNally, P.; McAuley, D.F.; Oglesby, I.K.; Wohlford-Lenane, C.L.; Bartlett, J.A.; Scott, C.J.; McElvaney, N.G.; Greene, C.M.; McCray, P.B.; et al. miR-31 dysregulation in cystic fibrosis airways contributes to increased pulmonary cathepsin S production. Am. J. Respir. Crit. Care Med. 2014, 190, 165–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.; Nath, S.; Lora, A.; Samaha, G.; Elgamal, Z.; Kaiser, R.; Taggart, C.; Weldon, S.; Geraghty, P. Cathepsin S: Investigating an old player in lung disease pathogenesis, comorbidities, and potential therapeutics. Respir. Res. 2020, 21, 111. [Google Scholar] [CrossRef] [PubMed]
- Velu, V.K.; Ramesh, R.; Srinivasan, A.R. Circulating MicroRNAs as Biomarkers in Health and Disease. J. Clin. Diagn. Res. 2012, 6, 1791–1795. [Google Scholar] [CrossRef]
- Wang, H.; Peng, R.; Wang, J.; Qin, Z.; Xue, L. Circulating microRNAs as potential cancer biomarkers: The advantage and disadvantage. Clin. Epigenetics 2018, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Russo, F.; Scoyni, F.; Fatica, A.; Pellegrini, M.; Ferro, A.; Pulvirenti, A.; Giugno, R. Chapter 12—Circulating Noncoding RNAs as Clinical Biomarkers. In Epigenetic Biomarkers and Diagnostics; García-Giménez, J.L., Ed.; Academic Press: Boston, MA, USA, 2016; pp. 239–258. ISBN 978-0-12-801899-6. [Google Scholar]
- Gibbings, D.J.; Ciaudo, C.; Erhardt, M.; Voinnet, O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat. Cell Biol. 2009, 11, 1143–1149. [Google Scholar] [CrossRef]
- Lim, S.B.; Di Lee, W.; Vasudevan, J.; Lim, W.-T.; Lim, C.T. Liquid biopsy: One cell at a time. NPJ Precis. Oncol. 2019, 3, 23. [Google Scholar] [CrossRef]
- Krause, K.; Kopp, B.T.; Tazi, M.F.; Caution, K.; Hamilton, K.; Badr, A.; Shrestha, C.; Tumin, D.; Hayes, D.; Robledo-Avila, F.; et al. The expression of Mirc1/Mir17–92 cluster in sputum samples correlates with pulmonary exacerbations in cystic fibrosis patients. J. Cyst. Fibros. 2018, 17, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Ideozu, J.E.; Zhang, X.; Rangaraj, V.; McColley, S.; Levy, H. Microarray profiling identifies extracellular circulating miRNAs dysregulated in cystic fibrosis. Sci. Rep. 2019, 9, 15483. [Google Scholar] [CrossRef] [Green Version]
- Chotirmall, S.H.; Greene, C.M.; Harvey, B.J.; McElvaney, N.G. The Cystic Fibrosis “Gender Gap”: Past Observations Present Understanding and Future Directions. In Cystic Fibrosis—Renewed Hopes Through Research; Sriramulu, D., Ed.; InTech: Vienna, Austria, 2012; ISBN 978-953-51-0287-8. [Google Scholar]
- Saint-Criq, V.; Harvey, B.J. Estrogen and the cystic fibrosis gender gap. Steroids 2014, 81, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Mooney, C.; McKiernan, P.J.; Raoof, R.; Henshall, D.C.; Linnane, B.; McNally, P.; Glasgow, A.M.A.; Greene, C.M. Plasma microRNA levels in male and female children with cystic fibrosis. Sci. Rep. 2020, 10, 1141. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.L.; Pereira, T.N.; Lewindon, P.J.; Shepherd, R.W.; Ramm, G.A. Circulating microRNAs as noninvasive diagnostic biomarkers of liver disease in children with cystic fibrosis. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Baldassarre, A.; Felli, C.; Prantera, G.; Masotti, A. Circulating microRNAs and Bioinformatics Tools to Discover Novel Diagnostic Biomarkers of Pediatric Diseases. Genes 2017, 8, 234. [Google Scholar] [CrossRef] [Green Version]
- Calvopina, D.A.; Chatfield, M.D.; Weis, A.; Coleman, M.A.; Fernandez-Rojo, M.A.; Noble, C.; Ramm, L.E.; Leung, D.H.; Lewindon, P.J.; Ramm, G.A. MicroRNA Sequencing Identifies a Serum MicroRNA Panel, Which Combined With Aspartate Aminotransferase to Platelet Ratio Index Can Detect and Monitor Liver Disease in Pediatric Cystic Fibrosis. Hepatology 2018, 68, 2301–2316. [Google Scholar] [CrossRef] [Green Version]
- Montanini, L.; Smerieri, A.; Gullì, M.; Cirillo, F.; Pisi, G.; Sartori, C.; Amarri, S.; Bernasconi, S.; Marmiroli, N.; Street, M.E. miR-146a, miR-155, miR-370, and miR-708 Are CFTR-Dependent, Predicted FOXO1 Regulators and Change at Onset of CFRDs. J. Clin. Endocrinol. Metab. 2016, 101, 4955–4963. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Pan, A.; Jia, S.; Ideozu, J.E.; Woods, K.; Murkowski, K.; Hessner, M.J.; Simpson, P.M.; Levy, H. Cystic Fibrosis Plasma Blunts the Immune Response to Bacterial Infection. Am. J. Respir. Cell Mol. Biol. 2019, 61, 301–311. [Google Scholar] [CrossRef]
- Staufer, K. Current Treatment Options for Cystic Fibrosis-Related Liver Disease. Int. J. Mol. Sci. 2020, 21, 8586. [Google Scholar] [CrossRef]
- Leeuwen, L.; Fitzgerald, D.A.; Gaskin, K.J. Liver disease in cystic fibrosis. Paediatr. Respir. Rev. 2014, 15, 69–74. [Google Scholar] [CrossRef]
- Betapudi, B.; Aleem, A.; Kothadia, J.P. Cystic Fibrosis and Liver Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Lewindon, P.J.; Shepherd, R.W.; Walsh, M.J.; Greer, R.M.; Williamson, R.; Pereira, T.N.; Frawley, K.; Bell, S.C.; Smith, J.L.; Ramm, G.A. Importance of hepatic fibrosis in cystic fibrosis and the predictive value of liver biopsy. Hepatology 2011, 53, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Ahanda, M.-L.E.; Bienvenu, T.; Sermet-Gaudelus, I.; Mazzolini, L.; Edelman, A.; Zoorob, R.; Davezac, N. The hsa-miR-125a/hsa-let-7e/hsa-miR-99b cluster is potentially implicated in Cystic Fibrosis pathogenesis. J. Cyst. Fibros. 2015, 14, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.-S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, J.F.; Cerqueira, L.; Figueiredo, C.; Oliveira, C.; Azevedo, N.F. Anti-miRNA oligonucleotides: A comprehensive guide for design. RNA Biol. 2018, 15, 338–352. [Google Scholar] [CrossRef] [Green Version]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef]
- Gumireddy, K.; Young, D.D.; Xiong, X.; Hogenesch, J.B.; Huang, Q.; Deiters, A. Small Molecule Inhibitors of MicroRNA miR-21 Function. Angew. Chem. Int. Ed. Engl. 2008, 47, 7482–7484. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.H.P.; Lim, S.; Wong, W.S.F. Antisense oligonucleotides: From design to therapeutic application. Clin. Exp. Pharm. Physiol. 2006, 33, 533–540. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Sikorski, A.F. Editorial focus: Understanding off-target effects as the key to successful RNAi therapy. Cell. Mol. Biol. Lett. 2019, 24, 69. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.; Sui, H.; Su, Y.; Cheng, W.; Liu, Y.; He, Z.; Ji, Q.; Xu, C. Research advances in molecular mechanisms underlying the pathogenesis of cystic fibrosis: From technical improvement to clinical applications (Review). Mol. Med. Rep. 2020, 22, 4992–5002. [Google Scholar] [CrossRef]
- Bardin, P.; Sonneville, F.; Corvol, H.; Tabary, O. Emerging microRNA Therapeutic Approaches for Cystic Fibrosis. Front. Pharm. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKiernan, P.J.; Cunningham, O.; Greene, C.M.; Cryan, S.-A. Targeting miRNA-based medicines to cystic fibrosis airway epithelial cells using nanotechnology. Int. J. Nanomed. 2013, 8, 3907–3915. [Google Scholar] [CrossRef] [Green Version]
- Amato, F.; Tomaiuolo, R.; Borbone, N.; Elce, A.; Amato, J.; D’Errico, S.; Rosa, G.D.; Mayol, L.; Piccialli, G.; Oliviero, G.; et al. Design, synthesis and biochemical investigation, by in vitro luciferase reporter system, of peptide nucleic acids as new inhibitors of miR-509-3p involved in the regulation of cystic fibrosis disease-gene expression. MedChemComm 2014, 5, 68–71. [Google Scholar] [CrossRef]
- Amato, F.; Tomaiuolo, R.; Nici, F.; Borbone, N.; Elce, A.; Catalanotti, B.; D’Errico, S.; Morgillo, C.M.; De Rosa, G.; Mayol, L.; et al. Exploitation of a Very Small Peptide Nucleic Acid as a New Inhibitor of miR-509-3p Involved in the Regulation of Cystic Fibrosis Disease-Gene Expression. Available online: https://www.hindawi.com/journals/bmri/2014/610718/ (accessed on 26 November 2020).
- Fabbri, E.; Tamanini, A.; Jakova, T.; Gasparello, J.; Manicardi, A.; Corradini, R.; Sabbioni, G.; Finotti, A.; Borgatti, M.; Lampronti, I.; et al. A Peptide Nucleic Acid against MicroRNA miR-145-5p Enhances the Expression of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) in Calu-3 Cells. Molecules 2017, 23, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbri, E.; Tamanini, A.; Jakova, T.; Gasparello, J.; Manicardi, A.; Corradini, R.; Finotti, A.; Borgatti, M.; Lampronti, I.; Munari, S.; et al. Treatment of human airway epithelial Calu-3 cells with a peptide-nucleic acid (PNA) targeting the microRNA miR-101-3p is associated with increased expression of the cystic fibrosis Transmembrane Conductance Regulator () gene. Eur. J. Med. Chem. 2020, 112876. [Google Scholar] [CrossRef]
- Sultan, S.; Rozzi, A.; Gasparello, J.; Manicardi, A.; Corradini, R.; Papi, C.; Finotti, A.; Lampronti, I.; Reali, E.; Cabrini, G.; et al. A Peptide Nucleic Acid (PNA) Masking the miR-145-5p Binding Site of the 3′UTR of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) mRNA Enhances CFTR Expression in Calu-3 Cells. Molecules 2020, 25, 1677. [Google Scholar] [CrossRef] [Green Version]
- Zarrilli, F.; Amato, F.; Morgillo, C.M.; Pinto, B.; Santarpia, G.; Borbone, N.; D’Errico, S.; Catalanotti, B.; Piccialli, G.; Castaldo, G.; et al. Peptide Nucleic Acids as miRNA Target Protectors for the Treatment of Cystic Fibrosis. Molecules 2017, 22, 1144. [Google Scholar] [CrossRef] [Green Version]
- Finotti, A.; Gasparello, J.; Fabbri, E.; Tamanini, A.; Corradini, R.; Dechecchi, M.C.; Cabrini, G.; Gambari, R. Enhancing the Expression of CFTR Using Antisense Molecules against MicroRNA miR-145-5p. Am. J. Respir. Crit. Care Med. 2019, 199, 1443–1444. [Google Scholar] [CrossRef]
- Santi, C.D.; Fernández, E.F.; Gaul, R.; Vencken, S.; Glasgow, A.; Oglesby, I.K.; Hurley, K.; Hawkins, F.; Mitash, N.; Mu, F.; et al. Precise Targeting of miRNA Sites Restores CFTR Activity in CF Bronchial Epithelial Cells. Mol. Ther. 2020, 28, 1190–1199. [Google Scholar] [CrossRef]
- McCarron, A.; Parsons, D.; Donnelley, M. Animal and Cell Culture Models for Cystic Fibrosis: Which Model Is Right for Your Application? Am. J. Pathol. 2020. [Google Scholar] [CrossRef]
- Semaniakou, A.; Croll, R.P.; Chappe, V. Animal Models in the Pathophysiology of Cystic Fibrosis. Front. Pharm. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakradhar, S. Put to the test: Organoid-based testing becomes a clinical tool. Nat. Med. 2017, 23, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.M.; Bijvelds, M.J.C.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Class of Mutation | CFTR Molecular Defect | Functional Abnormal Consequence | Mutation Examples | Type of Mutations | Clinical Phenotype | Therapeutic Strategy |
|---|---|---|---|---|---|---|
| I | No mRNA and protein synthesis | Absent protein | G542X, R553X, W1282X | Nonsense, frameshift, canonical splicing | Severe | Read-through agents |
| II | Reduced protein processing and traffic | Misfolded protein | F508del, N1303K, I507del | Missense, aminoacid deletion, | Severe | Correctors |
| III | Impaired channel gating | Reduced or absent channel opening | S549N, G551D | Missense, aminoacid change | Severe | Potentiators |
| IV | Decreased channel conductance | Defect in ion transport | R347P, R117H, D1152H | Missense, amino acid change | Mild | Potentiators |
| V | Reduced protein synthesis | Decreased protein | 3849 + 10 kb C>T, A455E | Splicing defect, missense | Mild | Potentiators, correctors, ASOs |
| VI | Less protein stability and protein turnover at cell surface | Decreased half-life of the protein | 120del23, G1412X | Missense, aminoacid change | Mild | Stabilisers |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Palma, F.D.E.; Raia, V.; Kroemer, G.; Maiuri, M.C. The Multifaceted Roles of MicroRNAs in Cystic Fibrosis. Diagnostics 2020, 10, 1102. https://doi.org/10.3390/diagnostics10121102
De Palma FDE, Raia V, Kroemer G, Maiuri MC. The Multifaceted Roles of MicroRNAs in Cystic Fibrosis. Diagnostics. 2020; 10(12):1102. https://doi.org/10.3390/diagnostics10121102
Chicago/Turabian StyleDe Palma, Fatima Domenica Elisa, Valeria Raia, Guido Kroemer, and Maria Chiara Maiuri. 2020. "The Multifaceted Roles of MicroRNAs in Cystic Fibrosis" Diagnostics 10, no. 12: 1102. https://doi.org/10.3390/diagnostics10121102
APA StyleDe Palma, F. D. E., Raia, V., Kroemer, G., & Maiuri, M. C. (2020). The Multifaceted Roles of MicroRNAs in Cystic Fibrosis. Diagnostics, 10(12), 1102. https://doi.org/10.3390/diagnostics10121102