Whole Exome Sequencing with Comprehensive Gene Set Analysis Identified a Biparental-Origin Homozygous c.509G>A Mutation in PPIB Gene Clustered in Two Taiwanese Families Exhibiting Fetal Skeletal Dysplasia during Prenatal Ultrasound

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
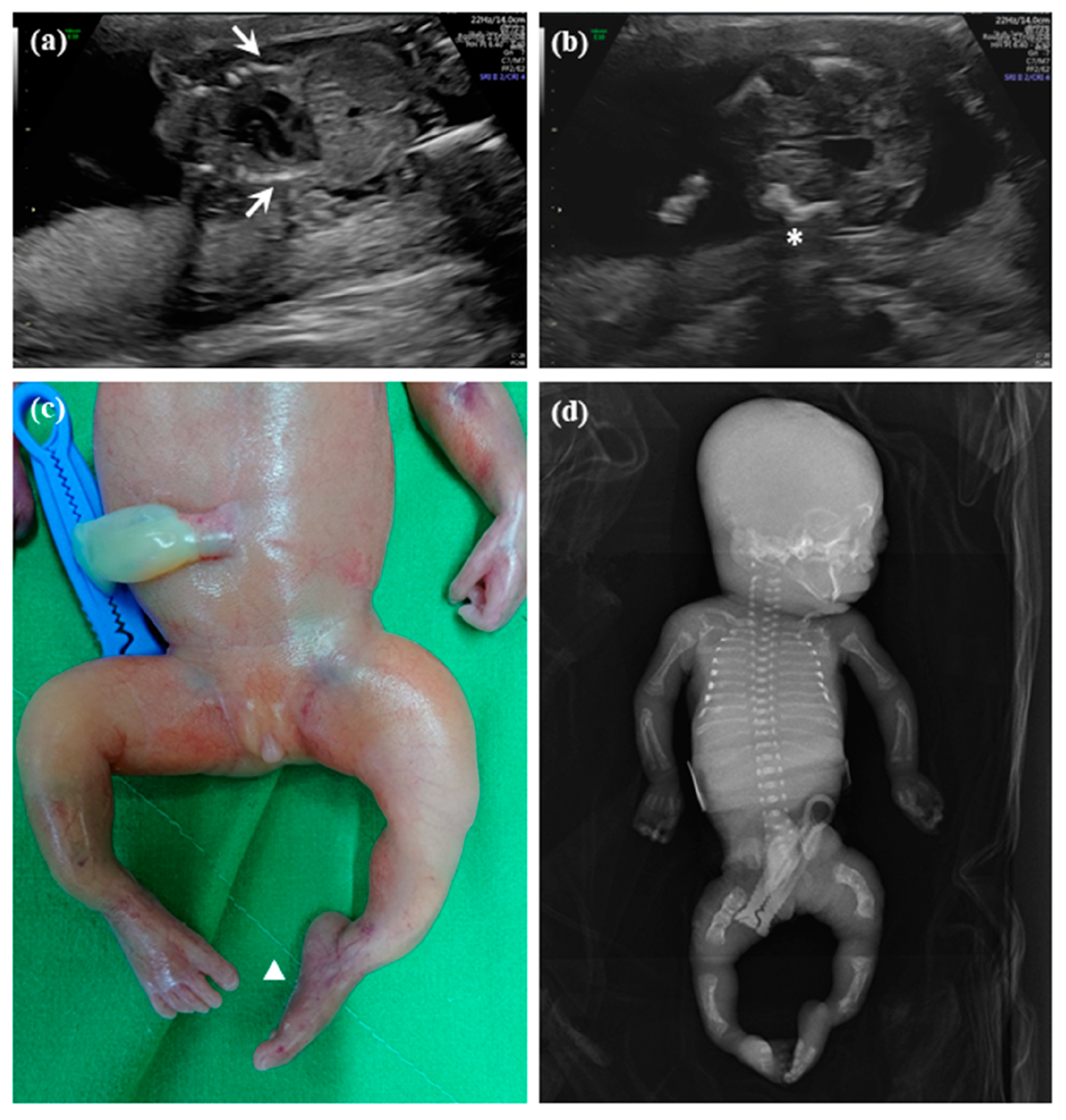
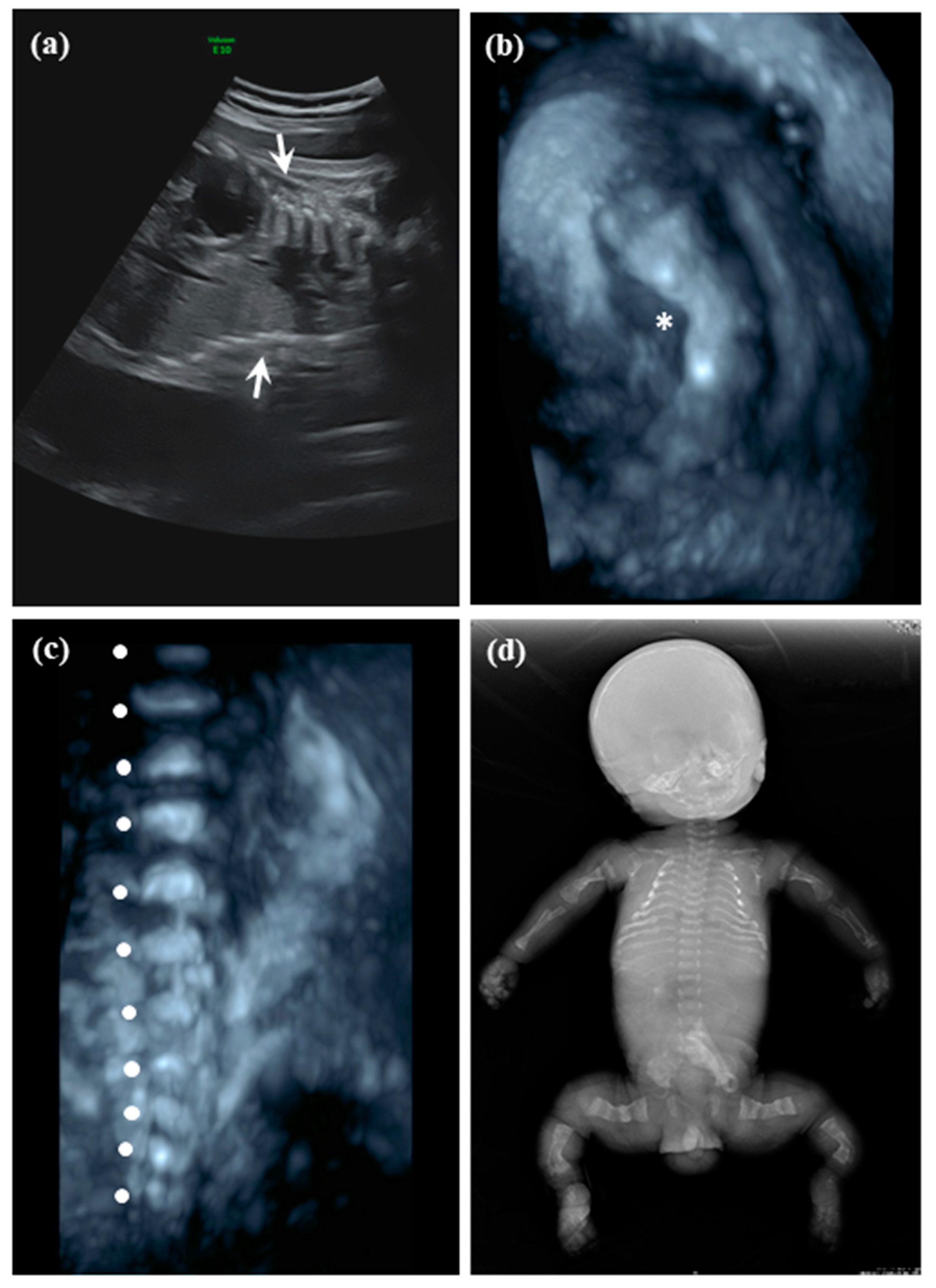
2.1.1. Patient 1
2.1.2. Patient 2
2.2. Whole Exome Sequencing (WES) by Next Generation Sequencing (NGS)
2.3. Exome Variation Analysis
2.4. In Silico Analysis
2.5. Cross-Species Conservation Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Milks, K.S.; Hill, L.M.; Hosseinzadeh, K. Evaluating skeletal dysplasias on prenatal ultrasound: An emphasis on predicting lethality. Pediatr Radiol. 2017, 47, 134–145. [Google Scholar] [CrossRef]
- Rimoin, D.L.; Cohn, D.; Krakow, D.; Wilcox, W.; Lachman, R.S.; Alanay, Y. The skeletal dysplasias: Clinical-molecular correlations. Ann. N. Y. Acad. Sci. 2007, 1117, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Victoria, T.; Zhu, X.; Lachman, R.; Epelman, M.; Oliver, E.R.; Adzick, N.S.; Biko, D.M. What Is New in Prenatal Skeletal Dysplasias? AJR Am. J. Roentgenol. 2018, 210, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Krakow, D.; Lachman, R.S.; Rimoin, D.L. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genet. Med. 2009, 11, 127–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marini, J.C.; Forlino, A.; Bachinger, H.P.; Bishop, N.J.; Byers, P.H.; Paepe, A.; Fassier, F.; Fratzl-Zelman, N.; Kozloff, K.M.; Krakow, D.; et al. Osteogenesis imperfecta. Nat. Rev. Dis. Primers 2017, 3, 17052. [Google Scholar] [CrossRef] [PubMed]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. A 2019, 179, 2393–2419. [Google Scholar] [CrossRef] [PubMed]
- Nikkel, S.M. Skeletal Dysplasias: What Every Bone Health Clinician Needs to Know. Curr. Osteoporos Rep. 2017, 15, 419–424. [Google Scholar] [CrossRef]
- Sa-Caputo, D.C.; Dionello, C.D.F.; Frederico, E.; Paineiras-Domingos, L.L.; Sousa-Goncalves, C.R.; Morel, D.S.; Moreira-Marconi, E.; Unger, M.; Bernardo-Filho, M. Whole-Body Vibration Exercise Improves Functional Parameters in Patients with Osteogenesis Imperfecta: A Systematic Review with a Suitable Approach. Afr. J. Tradit. Complement Altern. Med. 2017, 14, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, F.S.; Sillence, D.O. Osteogenesis imperfecta: Clinical diagnosis, nomenclature and severity assessment. Am. J. Med. Genet. A 2014, 164A, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Chandler, N.; Best, S.; Hayward, J.; Faravelli, F.; Mansour, S.; Kivuva, E.; Tapon, D.; Male, A.; DeVile, C.; Chitty, L.S. Rapid prenatal diagnosis using targeted exome sequencing: A cohort study to assess feasibility and potential impact on prenatal counseling and pregnancy management. Genet Med. 2018, 20, 1430–1437. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Day, I.N.; Gaunt, T.R. Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 2013, 29, 1504–1510. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Jhong, J.H.; Lee, J.; Koo, J.Y. Meta-analytic support vector machine for integrating multiple omics data. BioData Min. 2017, 10, 2. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.; Edwards, K.J.; Day, I.N.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef]
- Bonafe, L.; Cormier-Daire, V.; Hall, C.; Lachman, R.; Mortier, G.; Mundlos, S.; Nishimura, G.; Sangiorgi, L.; Savarirayan, R.; Sillence, D.; et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am. J. Med. Genet. A 2015, 167A, 2869–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyott, S.M.; Schwarze, U.; Christiansen, H.E.; Pepin, M.G.; Leistritz, D.F.; Dineen, R.; Harris, C.; Burton, B.K.; Angle, B.; Kim, K.; et al. Mutations in PPIB (cyclophilin B) delay type I procollagen chain association and result in perinatal lethal to moderate osteogenesis imperfecta phenotypes. Hum. Mol. Genet. 2011, 20, 1595–1609. [Google Scholar] [CrossRef] [Green Version]
- van Dijk, F.S.; Nesbitt, I.M.; Zwikstra, E.H.; Nikkels, P.G.; Piersma, S.R.; Fratantoni, S.A.; Jimenez, C.R.; Huizer, M.; Morsman, A.C.; Cobben, J.M.; et al. PPIB mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet. 2009, 85, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadares, E.R.; Carneiro, T.B.; Santos, P.M.; Oliveira, A.C.; Zabel, B. What is new in genetics and osteogenesis imperfecta classification? J. Pediatr (Rio. J.) 2014, 90, 536–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, R.H.; Hwu, W.L.; Chen, M.; Chung, I.F.; Peng, S.S.; Chen, C.Y.; Cheng, W.C.; Chien, Y.H.; Lee, N.C. Next-generation sequencing identifies TRPV4-related skeletal dysplasia in a boy with progressive bowlegs. Pediatr Neonatol. 2019, 60, 102–104. [Google Scholar] [CrossRef]
- Barnes, A.M.; Cabral, W.A.; Weis, M.; Makareeva, E.; Mertz, E.L.; Leikin, S.; Eyre, D.; Trujillo, C.; Marini, J.C. Absence of FKBP10 in recessive type XI osteogenesis imperfecta leads to diminished collagen cross-linking and reduced collagen deposition in extracellular matrix. Hum. Mutat. 2012, 33, 1589–1598. [Google Scholar] [CrossRef] [Green Version]
- Barnes, A.M.; Carter, E.M.; Cabral, W.A.; Weis, M.; Chang, W.; Makareeva, E.; Leikin, S.; Rotimi, C.N.; Eyre, D.R.; Raggio, C.L.; et al. Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N. Engl. J. Med. 2010, 362, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.M.; Nicholls, A.C.; Daw, S.C.; Mitchell, N.; Levin, L.S.; Green, B.; MacKenzie, J.; Evans, D.R.; Chudleigh, P.A.; Pope, F.M. Phenotypical features of an unique Irish family with severe autosomal recessive osteogenesis imperfecta. Clin. Genet. 1989, 35, 181–190. [Google Scholar] [CrossRef]
- Price, E.R.; Zydowsky, L.D.; Jin, M.J.; Baker, C.H.; McKeon, F.D.; Walsh, C.T. Human cyclophilin B: A second cyclophilin gene encodes a peptidyl-prolyl isomerase with a signal sequence. Proc. Natl. Acad. Sci. USA 1991, 88, 1903–1907. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Heitman, J. The cyclophilins. Genome Biol. 2005, 6, 226. [Google Scholar] [CrossRef] [Green Version]
- Schonbrunner, E.R.; Schmid, F.X. Peptidyl-prolyl cis-trans isomerase improves the efficiency of protein disulfide isomerase as a catalyst of protein folding. Proc. Natl. Acad. Sci. USA 1992, 89, 4510–4513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Pan, J.; Guo, D.; Zhang, W.; Xie, J.; Fang, Z.; Guo, C.; Fang, Q.; Jiang, W.; Guo, Y. Two novel mutations in the PPIB gene cause a rare pedigree of osteogenesis imperfecta type IX. Clin. Chim. Acta. 2017, 469, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.C.; Chien, Y.H.; Hwu, W.L. A review of aromatic l-amino acid decarboxylase (AADC) deficiency in Taiwan. Am. J. Med. Genet. C Semin Med. Genet. 2019, 181, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.C.; Chen, Y.C.; Wu, W.J.; Chang, S.P.; Chang, T.Y.; Lin, W.H.; Chen, M. Prenatal Diagnosis of Autosomal recessive renal tubular dysgenesis with anhydramnios caused by a mutation in the AGT gene. Diagnostics 2019, 9. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Zygosity (P/M/F) | Genomic Coordinate † | Gene | Associated Disease (Inheritance) | cDNA Change | Amino Acid Change | Type of Variation | dbSNP153 | Allele Frequency | ClinVar |
|---|---|---|---|---|---|---|---|---|---|---|
| Family 1 | ||||||||||
| homo/het/het | chr4 g.39270033A>G | WDR19 | 1.Cranioectodermal dysplasia 4; OMIM#614378 (AR) 2.Short-rib thoracic dysplasia 5; OMIM#614376 (AR) | NM_025132.4: c.3416A>G | p.Q1139R | Missense | rs75621037 | 0.00939 | Uncertain significance | |
| homo/het/het | chr12 g.88058860A>G | CEP290 | 1.Bardet-Biedl syndrome 14; OMIM#615991 (AR) 2.Joubert syndrome 5; OMIM#610188 (AR) 3.Meckel syndrome 4; OMIM#611134 (AR) 4.Senior-Loken syndrome 6; OMIM#610189 (AR) | NM_025114.4: c.6806T>C | p.I2269T | Missense | rs200090371 | 0.00114 | NA | |
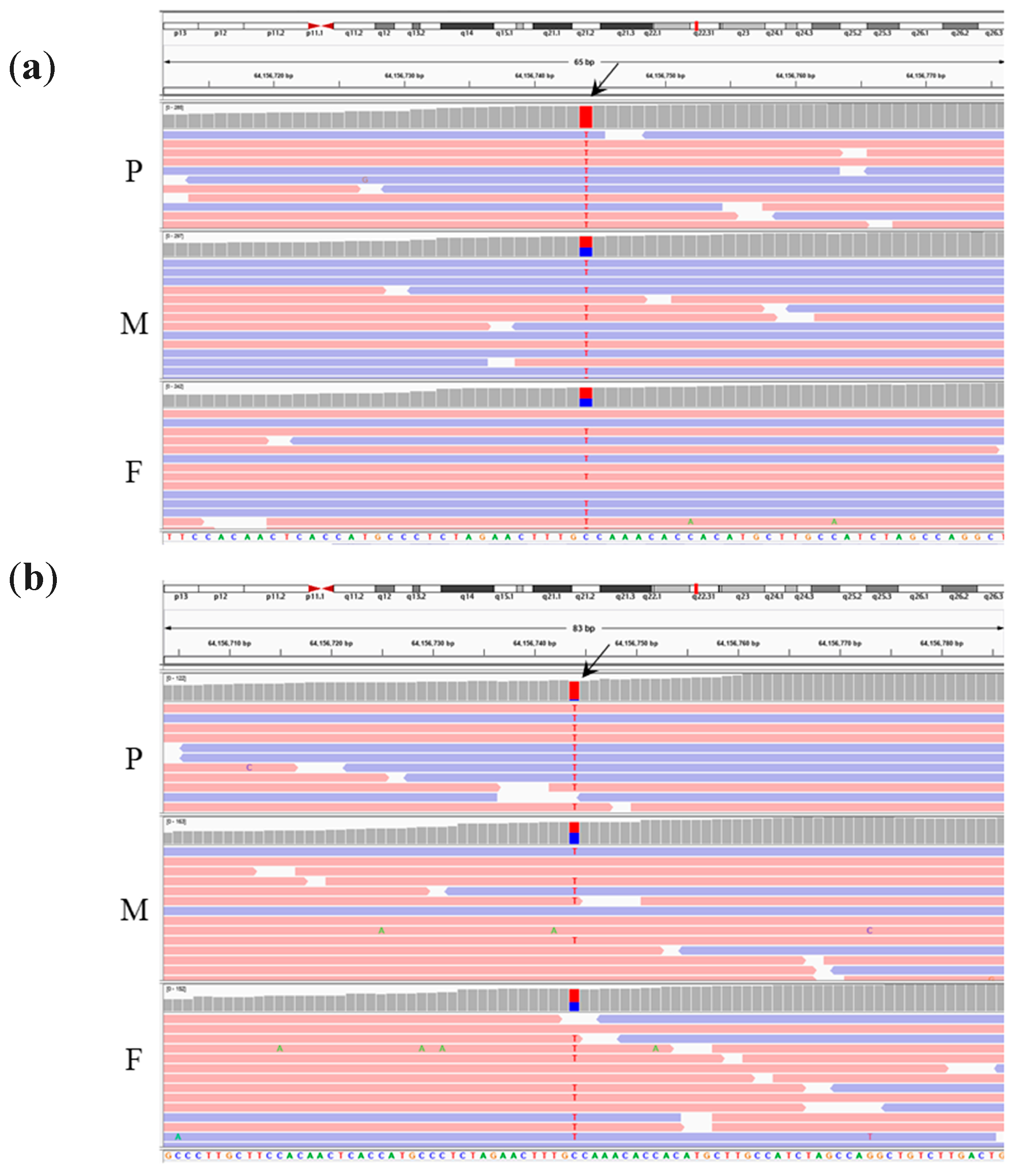
| homo/het/het | chr15 g.64156744C>T | PPIB | OI type IX; OMIM#259440 (AR) | NM_000942.5: c.509G>A | p.G170D | Missense | rs199606428 | 0.00035 | NA | |
| Family 2 | ||||||||||
| homo/het/het | chr15 g.64156744C>T | PPIB | OI type IX; OMIM#259440 (AR). | NM_000942.5: c.509G>A | p.G170D | Missense | rs199606428 | 0.00035 | NA |
| WDR19 c.3416A>G(p.Q1139R) | CEP290 c.6806T>C(p.I2269T) | PPIB c.509G>A(p.G170D) | |
|---|---|---|---|
| Reference transcript | NM_025132.4 | NM_025114.4 | NM_000942.5 |
| Prediction algorithm | |||
| SIFT | Tolerated | Tolerated | Deleterious |
| Polyphen 2 HVar | Benign | Benign | Damaging |
| LRT | Deleterious | Neutral | Deleterious |
| Mutation Taster | Disease causing | Polymorphism | Disease causing |
| Mutation Assessor † | Low | Low | High |
| FATHMM | Tolerated | Tolerated | Deleterious |
| PROVEAN | Neutral | Neutral | Deleterious |
| MetaSVM | Tolerated | Tolerated | Deleterious |
| MetaLR | Tolerated | Tolerated | Deleterious |
| FATHMM-MKL | Deleterious | Deleterious | Deleterious |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, T.-Y.; Chung, I.-F.; Wu, W.-J.; Chang, S.-P.; Lin, W.-H.; Ginsberg, N.A.; Ma, G.-C.; Chen, M. Whole Exome Sequencing with Comprehensive Gene Set Analysis Identified a Biparental-Origin Homozygous c.509G>A Mutation in PPIB Gene Clustered in Two Taiwanese Families Exhibiting Fetal Skeletal Dysplasia during Prenatal Ultrasound. Diagnostics 2020, 10, 286. https://doi.org/10.3390/diagnostics10050286
Chang T-Y, Chung I-F, Wu W-J, Chang S-P, Lin W-H, Ginsberg NA, Ma G-C, Chen M. Whole Exome Sequencing with Comprehensive Gene Set Analysis Identified a Biparental-Origin Homozygous c.509G>A Mutation in PPIB Gene Clustered in Two Taiwanese Families Exhibiting Fetal Skeletal Dysplasia during Prenatal Ultrasound. Diagnostics. 2020; 10(5):286. https://doi.org/10.3390/diagnostics10050286
Chicago/Turabian StyleChang, Ting-Yu, I-Fang Chung, Wan-Ju Wu, Shun-Ping Chang, Wen-Hsiang Lin, Norman A. Ginsberg, Gwo-Chin Ma, and Ming Chen. 2020. "Whole Exome Sequencing with Comprehensive Gene Set Analysis Identified a Biparental-Origin Homozygous c.509G>A Mutation in PPIB Gene Clustered in Two Taiwanese Families Exhibiting Fetal Skeletal Dysplasia during Prenatal Ultrasound" Diagnostics 10, no. 5: 286. https://doi.org/10.3390/diagnostics10050286
APA StyleChang, T. -Y., Chung, I. -F., Wu, W. -J., Chang, S. -P., Lin, W. -H., Ginsberg, N. A., Ma, G. -C., & Chen, M. (2020). Whole Exome Sequencing with Comprehensive Gene Set Analysis Identified a Biparental-Origin Homozygous c.509G>A Mutation in PPIB Gene Clustered in Two Taiwanese Families Exhibiting Fetal Skeletal Dysplasia during Prenatal Ultrasound. Diagnostics, 10(5), 286. https://doi.org/10.3390/diagnostics10050286