
Facilitations and Hurdles of Genetic Testing in Neuromuscular Disorders

Abstract
:1. Introduction
2. The Complexity of Diagnosing a Neuromuscular Disorder
3. The Approach to Genetic Testing
4. The Evolution of Genetic Techniques and Their Application to NMDs
5. NGS and Its Hurdles
5.1. GPS Panel Sequencing
5.2. Whole-Exome Sequencing (WES)
5.3. Whole-Genome Sequencing (WGS)
5.4. Mitochondrial Genome Sequencing
5.5. Data Analysis and Challenges
5.6. Emerging Technologies
6. Discussion
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zatz, M.; Passos-Bueno, M.R.; Vainzof, M. Neuromuscular disorders: Genes, genetic counseling and therapeutic trials. Genet. Mol. Biol. 2016, 39, 339–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, A.A.; Russel, J.A. Neuromuscular Disorders, 2nd ed.; McGraw-Hill Education: New York, NY, USA, 2016; pp. 2–21. [Google Scholar]
- Efthymiou, S.; Manole, A.; Houlden, H. Next-generation sequencing in neuromuscular diseases. Curr. Opin. Neurol. 2016, 29, 527–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Resta, C.; Pipitone, G.B.; Carrera, P.; Ferrari, M. Current scenario of the genetic testing for rare neurological disorders exploiting next generation sequencing. Neural. Regen. Res. 2021, 16, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Fogel, B.L. Genetic and genomic testing for neurologic disease in clinical practice. Handb. Clin. Neurol. 2018, 147, 11–22. [Google Scholar] [CrossRef]
- Toft, M. Advances in genetic diagnosis of neurological disorders. Acta Neurol. Scand. Suppl. 2014, 198, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Orengo, J.P.; Murdock, D.R. Genetic Testing in Neuromuscular Disorders. Understanding ordering and interpretation of genetic tests is paramount for clinical management. Pract. Neurol. 2019, 35–41. [Google Scholar]
- D’Amico, A.; Catteruccia, M.; Baranello, G.; Politano, L.; Govoni, A.; Previtali, S.C.; Pane, M.; D’Angelo, M.G.; Bruno, C.; Messina, S.; et al. Diagnosis of Duchenne Muscular Dystrophy in Italy in the last decade: Critical issues and areas for improvements. Neuromuscul. Disord. 2017, 27, 447–451. [Google Scholar] [CrossRef]
- Pera, M.C.; Coratti, G.; Berti, B.; D’Amico, A.; Sframeli, M.; Albamonte, E.; de Sanctis, R.; Messina, S.; Catteruccia, M.; Brigati, G.; et al. Diagnostic journey in Spinal Muscular Atrophy: Is it still an odyssey? PLoS ONE 2020, 15, e0230677. [Google Scholar] [CrossRef]
- Hilbert, J.E.; Johnson, N.E.; Moxley, R.T., 3rd. New insights about the incidence, multisystem manifestations, and care of patients with congenital myotonic dystrophy. J. Pediatr 2013, 163, 12–14. [Google Scholar] [CrossRef]
- Hilbert, J.E.; Ashizawa, T.; Day, J.W.; Luebbe, E.A.; Martens, W.B.; McDermott, M.P.; Tawil, R.; Thornton, C.A.; Moxley, R.T., 3rd. Diagnostic odyssey of patients with myotonic dystrophy. J. Neurol. 2013, 260, 2497–2504. [Google Scholar] [CrossRef] [Green Version]
- Zampatti, S.; Colantoni, L.; Strafella, C.; Galota, R.M.; Caputo, V.; Campoli, G.; Pagliaroli, G.; Carboni, S.; Mela, J.; Peconi, C.; et al. Facioscapulohumeral muscular dystrophy (FSHD) molecular diagnosis: From traditional technology to the NGS era. Neurogenetics 2019, 20, 57–64. [Google Scholar] [CrossRef]
- Martínez-Molina, M.; Argente-Escrig, H.; Polo, M.F.; Hervás, D.; Frasquet, M.; Cortés, V.; Sevilla, T.; Vázquez-Costa, J.F. Early Referral to an ALS Center Reduces Several Months the Diagnostic Delay: A Multicenter-Based Study. Front. Neurol. 2020, 11, 604922. [Google Scholar] [CrossRef]
- Peterlin, B.; Gualandi, F.; Maver, A.; Servidei, S.; van der Maarel, S.M.; Lamy, F.; Mejat, A.; Evangelista, T.; Ferlini, A. Genetic testing offer for inherited neuromuscular diseases within the EURO-NMD reference network: A European survey study. PLoS ONE 2020, 15, e0239329. [Google Scholar] [CrossRef]
- Arnold, W.D.; Flanigan, K.M. A practical approach to molecular diagnostic testing in neuromuscular diseases. Phys. Med. Rehabil. Clin. N. Am. 2012, 23, 589–608. [Google Scholar] [CrossRef]
- Vgontzas, A.; Renthal, W. Introduction to neurogenetics. Am. J. Med. 2019, 132, 142–152. [Google Scholar] [CrossRef]
- Vivekanandam, V.; Männikkö, R.; Matthews, E.; Hanna, M.G. Improving genetic diagnostics of skeletal muscle channelopathies. Expert Rev. Mol. Diagn. 2020, 20, 725–736. [Google Scholar] [CrossRef]
- Durmaz, A.A.; Karac, E.; Demkow, U.; Toruner, G.; Schoumans, J.; Cogulu, O. Evolution of Genetic Techniques: Past, Present, and Beyond. Biomed. Res. Int. 2015, 2015, 461524. [Google Scholar] [CrossRef]
- Watson, J.D.; Crick, F.H. The structure of DNA. Cold Spring Harb. Symp Quant. Biol. 1953, 18, 123–131. [Google Scholar] [CrossRef]
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific enzymatic amplification of DNA in vitro: The polymerase chain reaction. Cold Spring Harb. Symp Quant. Biol. 1986, 51, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [Green Version]
- Marian, A.J. Clinical Interpretation and Management of Genetic Variants. JACC Basic Transl. Sci. 2020, 5, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Shieh, P.B. Advances in the Genetic Testing of Neuromuscular Diseases. Neurol. Clin. 2020, 38, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Volk, A.E.; Kubisch, C. The rapid evolution of molecular genetic diagnostics in neuromuscular diseases. Curr. Opin. Neurol. 2017, 30, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.; Spendiff, S.; Roos, A.; Bourque, P.R.; Warman Chardon, J.; Kirschner, J.; Horvath, R.; Lochmüller, H. Advances in the diagnosis of inherited neuromuscular diseases and implications for therapy development. Lancet Neurol. 2020, 19, 522–532. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Shatunov, A.; Al-Chalabi, A. The genetic architecture of ALS. Neurobiol. Dis. 2021, 147, 105156. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Deignan, J.L.; Dorrani, N.; Strom, S.P.; Kantarci, S.; Quintero-Rivera, F.; Das, K.; Toy, T.; Harry, B.; Yourshaw, M.; et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 2014, 312, 1880–1887. [Google Scholar] [CrossRef]
- Yavarna, T.; Al-Dewik, N.; Al-Mureikhi, M.; Ali, R.; Al-Mesaifri, F.; Mahmoud, L.; Shahbeck, N.; Lakhani, S.; AlMulla, M.; Nawaz, Z.; et al. High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum. Genet. 2015, 134, 967–980. [Google Scholar] [CrossRef]
- Ankala, A.; da Silva, C.; Gualandi, F.; Ferlini, A.; Bean, L.J.; Collins, C.; Tanner, A.K.; Hegde, M.R. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann. Neurol. 2015, 77, 206–214. [Google Scholar] [CrossRef]
- Klein, C.J.; Middha, S.; Duan, X.; Wu, Y.; Litchy, W.J.; Gu, W.; Dyck, P.J.; Gavrilova, R.H.; Smith, D.I.; Kocher, J.P.; et al. Application of whole exome sequencing in undiagnosed inherited polyneuropathies. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1265–1272. [Google Scholar] [CrossRef]
- Chae, J.H.; Vasta, V.; Cho, A.; Lim, B.C.; Zhang, Q.; Eun, S.H.; Hahn, S.H. Utility of next generation sequencing in genetic diagnosis of early onset neuromuscular disorders. J. Med. Genet. 2015, 52, 208–216. [Google Scholar] [CrossRef]
- Ghaoui, R.; Cooper, S.T.; Lek, M.; Jones, K.; Corbett, A.; Reddel, S.W.; Needham, M.; Liang, C.; Waddell, L.B.; Nicholson, G.; et al. Use of Whole-Exome Sequencing for Diagnosis of Limb-Girdle Muscular Dystrophy: Outcomes and Lessons Learned. JAMA Neurol. 2015, 72, 1424–1432. [Google Scholar] [CrossRef]
- Gorokhova, S.; Cerino, M.; Mathieu, Y.; Courrier, S.; Desvignes, J.P.; Salgado, D.; Béroud, C.; Krahn, M.; Bartoli, M. Comparing targeted exome and whole exome approaches for genetic diagnosis of neuromuscular disorders. Appl. Transl. Genom. 2015, 7, 26–31. [Google Scholar] [CrossRef]
- Tian, X.; Liang, W.C.; Feng, Y.; Wang, J.; Zhang, V.W.; Chou, C.H.; Huang, H.D.; Lam, C.W.; Hsu, Y.Y.; Lin, T.S.; et al. Expanding genotype/phenotype of neuromuscular diseases by comprehensive target capture/NGS. Neurol. Genet. 2015, 1, e14. [Google Scholar] [CrossRef] [Green Version]
- Evilä, A.; Arumilli, M.; Udd, B.; Hackman, P. Targeted next-generation sequencing assay for detection of mutations in primary myopathies. Neuromuscul. Disord. 2016, 26, 7–15. [Google Scholar] [CrossRef]
- Fattahi, Z.; Kalhor, Z.; Fadaee, M.; Vazehan, R.; Parsimehr, E.; Abolhassani, A.; Beheshtian, M.; Zamani, G.; Nafissi, S.; Nilipour, Y.; et al. Improved diagnostic yield of neuromuscular disorders applying clinical exome sequencing in patients arising from a consanguineous population. Clin. Genet. 2017, 91, 386–402. [Google Scholar] [CrossRef]
- Haskell, G.T.; Adams, M.C.; Fan, Z.; Amin, K.; Guzman Badillo, R.J.; Zhou, L.; Bizon, C.; Chahin, N.; Greenwood, R.S.; Milko, L.V.; et al. Diagnostic utility of exome sequencing in the evaluation of neuromuscular disorders. Neurol. Genet. 2018, 4, e212. [Google Scholar] [CrossRef] [Green Version]
- Schofield, D.; Alam, K.; Douglas, L.; Shrestha, R.; MacArthur, D.G.; Davis, M.; Laing, N.G.; Clarke, N.F.; Burns, J.; Cooper, S.T.; et al. Cost-effectiveness of massively parallel sequencing for diagnosis of paediatric muscle diseases. NPJ Genom. Med. 2017, 2, 4. [Google Scholar] [CrossRef]
- Koboldt, D.C. Best practices for variant calling in clinical sequencing. Genome. Med. 2020, 12, 91. [Google Scholar] [CrossRef]
- Strande, N.T.; Brnich, S.E.; Roman, T.S.; Berg, J.S. Navigating the nuances of clinical sequence variant interpretation in Mendelian disease. Genet. Med. 2018, 20, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Beecroft, S.J.; Yau, K.S.; Allcock, R.J.N.; Mina, K.; Gooding, R.; Faiz, F.; Atkinson, V.J.; Wise, C.; Sivadorai, P.; Trajanoski, D.; et al. Targeted gene panel use in 2249 neuromuscular patients: The Australasian referral center experience. Ann. Clin. Transl. Neurol. 2020, 7, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Marmiesse, A.; Gouveia, S.; Couce, M.L. NGS Technologies as a Turning Point in Rare Disease Research, Diagnosis and Treatment. Curr. Med. Chem. 2018, 25, 404–432. [Google Scholar] [CrossRef]
- Todd, E.J.; Yau, K.S.; Ong, R.; Slee, J.; McGillivray, G.; Barnett, C.P.; Haliloglu, G.; Talim, B.; Akcoren, Z.; Kariminejad, A.; et al. Next generation sequencing in a large cohort of patients presenting with neuromuscular disease before or at birth. Orphanet. J. Rare Dis. 2015, 10, 148. [Google Scholar] [CrossRef] [Green Version]
- Lévesque, S.; Auray-Blais, C.; Gravel, E.; Boutin, M.; Dempsey-Nunez, L.; Jacques, P.E.; Chenier, S.; Larue, S.; Rioux, M.F.; Al-Hertani, W.; et al. Diagnosis of late-onset Pompe disease and other muscle disorders by next-generation sequencing. Orphanet J. Rare Dis. 2016, 11, 8. [Google Scholar] [CrossRef]
- Brugnoni, R.; Maggi, L.; Canioni, E.; Verde, F.; Gallone, A.; Ariattik, A.; Filosto, M.; Petrelli, C.; Logullo, F.O.; Esposito, M.; et al. Next-generation sequencing application to investigate skeletal muscle channelopathies in a large cohort of Italian patients. Neuromuscul. Disord. 2020. [Google Scholar] [CrossRef]
- Lamp, M.; Origone, P.; Geroldi, A.; Verdiani, S.; Gotta, F.; Caponnetto, C.; Devigili, G.; Verriello, L.; Scialò, C.; Cabona, C.; et al. Twenty years of molecular analyses in amyotrophic lateral sclerosis: Genetic landscape of Italian patients. Neurobiol. Aging 2018, 66, 179.e5–179.e16. [Google Scholar] [CrossRef]
- Montenegro, G.; Powell, E.; Huang, J.; Speziani, F.; Edwards, Y.J.; Beecham, G.; Hulme, W.; Siskind, C.; Vance, J.; Shy, M.; et al. Exome sequencing allows for rapid gene identification in a Charcot-Marie-Tooth family. Ann. Neurol. 2011, 69, 464–470. [Google Scholar] [CrossRef] [Green Version]
- Waldrop, M.A.; Pastore, M.; Schrader, R.; Sites, E.; Bartholomew, D.; Tsao, C.Y.; Flanigan, K.M. Diagnostic Utility of Whole Exome Sequencing in the Neuromuscular Clinic. Neuropediatrics 2019, 50, 96–102. [Google Scholar] [CrossRef]
- LaDuca, H.; Farwell, K.D.; Vuong, H.; Lu, H.M.; Mu, W.; Shahmirzadi, L.; Tang, S.; Chen, J.; Bhide, S.; Chao, E.C. Exome sequencing covers >98% of mutations identified on targeted next generation sequencing panels. PLoS ONE 2017, 12, e0170843. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.S.; Fredrich, B.; Hoeppner, M.P.; Ellinghaus, D.; Franke, A. Opportunities and challenges of whole-genome and -exome sequencing. BMC Genet. 2017, 18, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shashi, V.; McConkie-Rosell, A.; Schoch, K.; Kasturi, V.; Rehder, C.; Jiang, Y.H.; Goldstein, D.B.; McDonald, M.T. Practical considerations in the clinical application of whole-exome sequencing. Clin. Genet. 2016, 89, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, D.; Heeley, J.; Vineyard, M.; Manwaring, L.; Toler, T.L.; Fassi, E.; Fiala, E.; Brown, S.; Goss, C.W.; Willing, M.; et al. The Exome Clinic and the role of medical genetics expertise in the interpretation of exome sequencing results. Genet. Med. 2017, 19, 1040–1048. [Google Scholar] [CrossRef] [Green Version]
- Tsai, G.J.; Rañola, J.M.O.; Smith, C.; Garrett, L.T.; Bergquist, T.; Casadei, S.; Bowen, D.J.; Shirts, B.H. Outcomes of 92 patient-driven family studies for reclassification of variants of uncertain significance. Genet. Med. 2019, 21, 1435–1442. [Google Scholar] [CrossRef]
- Ravenscroft, G.; Laing, N.G.; Bönnemann, C.G. Pathophysiological concepts in the congenital myopathies: Blurring the boundaries, sharpening the focus. Brain 2015, 138, 246–268. [Google Scholar] [CrossRef] [Green Version]
- Oates, E.C.; Jones, K.J.; Donkervoort, S.; Charlton, A.; Brammah, S.; Smith, J.E., 3rd; Ware, J.S.; Yau, K.S.; Swanson, L.C.; Whiffin, N.; et al. Congenital Titinopathy: Comprehensive characterization and pathogenic insights. Ann. Neurol. 2018, 83, 1105–1124. [Google Scholar] [CrossRef] [Green Version]
- Jungbluth, H.; Dowling, J.J.; Ferreiro, A.; Muntoni, F. RYR1 Myopathy Consortium. 217th ENMC International Workshop: RYR1-related myopathies, Naarden, The Netherlands, 29–31 January 2016. Neuromuscul. Disord. 2016, 26, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Mazzarotto, F.; Olivotto, I.; Walsh, R. Advantages and Perils of Clinical Whole-Exome and Whole-Genome Sequencing in Cardiomyopathy. Cardiovasc. Drugs 2020, 34, 241–253. [Google Scholar] [CrossRef]
- Lupski, J.R.; Reid, J.G.; Gonzaga-Jauregui, C.; Rio Deiros, D.; Chen, D.C.; Nazareth, L.; Bainbridge, M.; Dinh, H.; Jing, C.; Wheeler, D.A.; et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N. Engl. J. Med. 2010, 362, 1181–1191. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Kim, C.; Bradfield, J.; Guo, Y.; Toskala, E.; Otieno, F.G.; Hou, C.; Thomas, K.; Cardinale, C.; Lyon, G.J.; et al. Whole-genome DNA/RNA sequencing identifies truncating mutations in RBCK1 in a novel Mendelian disease with neuromuscular and cardiac involvement. Genome. Med. 2013, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Huang, Y.; Nie, Y.; Li, J.; Chen, G.; Tu, S.; Shen, P.; Chen, C. A novel PMP22 insertion mutation causing Charcot-Marie-Tooth disease type 3: A case report. Medicine 2021, 100, e25163. [Google Scholar] [CrossRef] [PubMed]
- Petrikin, J.E.; Willig, L.K.; Smith, L.D.; Kingsmore, S.F. Rapid whole genome sequencing and precision neonatology. Semin Perinatol. 2015, 39, 623–631. [Google Scholar] [CrossRef] [Green Version]
- Kremer, L.S.; Wortmann, S.B.; Prokisch, H. “Transcriptomics”: Molecular diagnosis of inborn errors of metabolism via RNA-sequencing. J. Inherit. Metab. Dis. 2018, 41, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Alfares, A.; Aloraini, T.; Al Subaie, L.; Alissa, A.; Al Qudsi, A.; Alahmad, A.; Al Mutairi, F.; Alswaid, A.; Alothaim, A.; Eyaid, W.; et al. Whole-genome sequencing offers additional but limited clinical utility compared with reanalysis of whole-exome sequencing. Genet. Med. 2018, 20, 1328–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schon, K.R.; Ratnaike, T.; van den Ameele, J.; Horvath, R.; Chinnery, P.F. Mitochondrial Diseases: A Diagnostic Revolution. Trends Genet. 2020, 36, 702–717. [Google Scholar] [CrossRef]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J., 2nd; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef]
- Neveling, K.; Feenstra, I.; Gilissen, C.; Hoefsloot, L.H.; Kamsteeg, E.J.; Mensenkamp, A.R.; Rodenburg, R.J.; Yntema, H.G.; Spruijt, L.; Vermeer, S.; et al. A post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum. Mutat. 2013, 34, 1721–1726. [Google Scholar] [CrossRef]
- Frazier, A.E.; Thorburn, D.R.; Compton, A. G Mitochondrial energy generation disorders: Genes, mechanisms, and clues to pathology. J. Biol. Chem. 2019, 294, 5386–5395. [Google Scholar] [CrossRef] [Green Version]
- Gusic, M.; Prokisch, H. Genetic basis of mitochondrial diseases. FEBS Lett. 2021. [Google Scholar] [CrossRef]
- Tang, S.; Wang, J.; Zhang, V.W.; Li, F.Y.; Landsverk, M.; Cui, H.; Truong, C.K.; Wang, G.; Chen, L.C.; Graham, B.; et al. Transition to next generation analysis of the whole mitochondrial genome: A summary of molecular defects. Hum. Mutat. 2013, 34, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Tucker, E.J.; Compton, A.G.; Kirby, D.M.; Crawford, G.; Burtt, N.P.; Rivas, M.; Guiducci, C.; Bruno, D.L.; Goldberger, O.A.; et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat. Genet. 2010, 42, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Vasta, V.; Ng, S.B.; Turner, E.H.; Shendure, J.; Hahn, S.H. Next generation sequence analysis for mitochondrial disorders. Genome Med. 2009, 1, 100. [Google Scholar] [CrossRef] [Green Version]
- DaRe, J.T.; Vasta, V.; Penn, J.; Tran, N.T.B.; Hahn, S.H. Targeted exome sequencing for mitochondrial disorders reveals high genetic heterogeneity. BMC Med. Genet. 2013, 14, 118. [Google Scholar] [CrossRef] [Green Version]
- Legati, A.; Reyes, A.; Nasca, A.; Invernizzi, F.; Lamantea, E.; Tiranti, V.; Garavaglia, B.; Lamperti, C.; Ardissone, A.; Moroni, I.; et al. New genes and pathomechanisms in mitochondrial disorders unravelled by NGS technologies. BBA-Bioenergetics 2016, 1857, 1326–1335. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Mayr, J.A.; Nuoffer, J.M.; Prokisch, H.; Sperl, W. A guideline for the diagnosis of pediatric mitochondrial disease: The value of muscle and skin biopsies in the genetics era. Neuropediatrics 2017, 48, 309–314. [Google Scholar] [CrossRef]
- Wagner, M.; Berutti, R.; Lorenz-Depiereux, B.; Graf, E.; Eckstein, G.; Mayr, J.A.; Meitinger, T.; Ahting, U.; Prokisch, H.; Strom, T.M.; et al. Mitochondrial DNA mutation analysis from exome sequencing-A more holistic approach in diagnostics of suspected mitochondrial disease. J. Inherit. Metab. Dis. 2019, 42, 909–917. [Google Scholar] [CrossRef]
- Wolf, N.I.; Smeitink, J.A. Mitochondrial disorders: A proposal for consensus diagnostic criteria in infants and children. Neurology 2002, 59, 1402–1405. [Google Scholar] [CrossRef]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Ciara, E.; Trubicka, J.; Rokicki, D.; Karkucinska-Wieckowska, A.; Pajdowska, M.; Jurkiewicz, E.; Halat, P.; Kosinska, J.; et al. New perspective in diagnostics of mitochondrial disorders: Two years’ experience with whole-exome sequencing at a national paediatric centre. J. Transl. Med. 2016, 14, 174. [Google Scholar] [CrossRef] [Green Version]
- Puusepp, S.; Reinson, K.; Pajusalu, S.; Murumets, U.; Oiglane-Shlik, E.; Rein, R.; Talvik, I.; Rodenburg, R.J.; Ounap, K. Effectiveness of whole exome sequencing in unsolved patients with a clinical suspicion of a mitochondrial disorder in Estonia. Mol. Genet. Metab. Rep. 2018, 15, 80–89. [Google Scholar] [CrossRef]
- Theunissen, T.E.J.; Nguyen, M.; Kamps, R.; Hendrickx, A.T.; Sallevelt, S.; Gottschalk, R.W.H.; Calis, C.M.; Stassen, A.P.M.; de Koning, B.; Mulder-Den Hartog, E.N.M.; et al. Whole exome sequencing is the preferred strategy to identify the genetic defect in patients with a probable or possible mitochondrial cause. Front. Genet. 2018, 9, 400. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.G.; Cowley, M.J.; Gayevskiy, V.; Minoche, A.E.; Puttick, C.; Thorburn, D.R.; Rius, R.; Compton, A.G.; Menezes, M.J.; Bhattacharya, K.; et al. The diagnostic utility of genome sequencing in a pediatric cohort with suspected mitochondrial disease. Genet. Med. 2020, 22, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Yu, Y.; Qing, T.; Guo, L.; Shi, L. Next-generation sequencing in the clinic: Promises and challenges. Cancer Lett. 2013, 342, 284–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, W.; Lu, H.M.; Chen, J.; Li, S.; Elliott, A.M. Sanger Confirmation Is Required to Achieve Optimal Sensitivity and Specificity in Next-Generation Sequencing Panel Testing. J. Mol. Diagn. 2016, 18, 923–932. [Google Scholar] [CrossRef] [Green Version]
- Bobo, D.; Lipatov, M.; Rodriguez-Flores, J.L.; Auton, A.; Henn, B.M. False Negatives Are a Significant Feature of Next Generation Sequencing Callsets. Biorxiv 2016. [Google Scholar] [CrossRef] [Green Version]
- Wenger, A.M.; Guturu, H.; Bernstein, J.A.; Bejerano, G. Systematic reanalysis of clinical exome data yields additional diagnoses: Implications for providers. Genet. Med. 2017, 19, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021. [Google Scholar] [CrossRef]
- Mitsuhashi, S.; Nakagawa, S.; Takahashi Ueda, M.; Imanishi, T.; Frith, M.C.; Mitsuhashi, H. Nanopore-based single molecule sequencing of the D4Z4 array responsible for facioscapulohumeral muscular dystrophy. Sci. Rep. 2017, 7, 14789. [Google Scholar] [CrossRef] [Green Version]
- Ebbert, M.T.W.; Farrugia, S.L.; Sens, J.P.; Jansen-West, K.; Gendron, T.F.; Prudencio, M.; McLaughlin, I.J.; Bowman, B.; Seetin, M.; DeJesus-Hernandez, M.; et al. Long-read sequencing across the C9orf72 ‘GGGGCC’ repeat expansion: Implications for clinical use and genetic discovery efforts in human disease. Mol. Neurodegener. 2018, 13, 46. [Google Scholar] [CrossRef] [Green Version]
- Giesselmann, P.; Brändl, B.; Raimondeau, E.; Bowen, R.; Rohrandt, C.; Tandon, R.; Kretzmer, H.; Assum, G.; Galonska, C.; Siebert, R.; et al. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat. Biotechnol. 2019, 37, 1478–1481. [Google Scholar] [CrossRef] [Green Version]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Van der Ende, E.L.; Jackson, J.L.; White, A.; Seelaar, H.; van Blitterswijk, M.; Van Swieten, J.C. Unravelling the clinical spectrum and the role of repeat length in C9ORF72 repeat expansions. J. Neurol. Neurosurg. Psychiatry 2021. [Google Scholar] [CrossRef]
- Ji, F.; Sadreyev, R.I. RNA-seq: Basic Bioinformatics Analysis. Curr. Protoc. Mol. Biol. 2018, 124, e68. [Google Scholar] [CrossRef]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef]
- Cummings, B.B.; Marshall, J.L.; Tukiainen, T.; Lek, M.; Donkervoort, S.; Foley, A.R.; Bolduc, V.; Waddell, L.B.; Sandaradura, S.A.; O’Grady, G.L.; et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med. 2017, 9, eaal5209. [Google Scholar] [CrossRef] [Green Version]
- Bolduc, V.; Foley, A.R.; Solomon-Degefa, H.; Sarathy, A.; Donkervoort, S.; Hu, Y.; Chen, G.S.; Sizov, K.; Nalls, M.; Zhou, H.; et al. A recurrent COL6A1 pseudoexon insertion causes muscular dystrophy and is effectively targeted by splice-correction therapies. JCI Insight 2019, 4, e124403. [Google Scholar] [CrossRef] [Green Version]
- Kremer, L.S.; Bader, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun. 2017, 8, 15824. [Google Scholar] [CrossRef]
- Gonorazky, H.D.; Naumenko, S.; Ramani, A.K.; Nelakuditi, V.; Mashouri, P.; Wang, P.; Kao, D.; Ohri, K.; Viththiyapaskaran, S.; Tarnopolsky, M.A.; et al. Expanding the Boundaries of RNA Sequencing as a Diagnostic Tool for Rare Mendelian Disease. Am. J. Hum. Genet. 2019, 104, 1007. [Google Scholar] [CrossRef] [Green Version]
- Kernohan, K.D.; Frésard, L.; Zappala, Z.; Hartley, T.; Smith, K.S.; Wagner, J.; Xu, H.; McBride, A.; Bourque, P.R.; Consortium, C.R.C.; et al. Whole-transcriptome sequencing in blood provides a diagnosis of spinal muscular atrophy with progressive myoclonic epilepsy. Hum. Mutat. 2017, 38, 611–614. [Google Scholar] [CrossRef]
- Elsaid, M.F.; Chalhoub, N.; Ben-Omran, T.; Kumar, P.; Kamel, H.; Ibrahim, K.; Mohamoud, Y.; Al-Dous, E.; Al-Azwani, I.; Malek, J.A.; et al. Mutation in noncoding RNA RNU12 causes early onset cerebellar ataxia. Ann. Neurol. 2017, 81, 68–78. [Google Scholar] [CrossRef]
- Kassardjian, C.D.; Amato, A.A.; Boon, A.J.; Boon, A.J.; Childers, M.K.; Klein, C.J.; AANEM Professional Practice Committee. The utility of genetic testing in neuromuscular disease: A consensus statement from the AANEM on the clinical utility of genetic testing in diagnosis of neuromuscular disease. Muscle Nerve 2016, 54, 1007–1009. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Ravi, B.; Antonellis, A.; Sumner, C.J.; Lieberman, A.P. Genetic approaches to the treatment of inherited neuromuscular diseases. Hum. Mol. Genet. 2019, 28, R55–R64. [Google Scholar] [CrossRef] [Green Version]
- Roggenbuck, J.; Quick, A.; Kolb, S.J. Genetic testing and genetic counseling for amyotrophic lateral sclerosis: An update for clinicians. Genet. Med. 2017, 19, 267–274. [Google Scholar] [CrossRef] [Green Version]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.J.; Hwu, W.L.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul. Disord. 2019, 29, 842–856. [Google Scholar] [CrossRef] [Green Version]
- Pane, M.; Coratti, G.; Sansone, V.A.; Messina, S.; Catteruccia, M.; Bruno, C.; Sframeli, M.; Albamonte, E.; Pedemonte, M.; D’Amico, A.; et al. Type I SMA “new natural history”: Long-term data in nusinersen-treated patients. Ann. Clin. Transl. Neurol. 2021, 8, 548–557. [Google Scholar] [CrossRef] [PubMed]



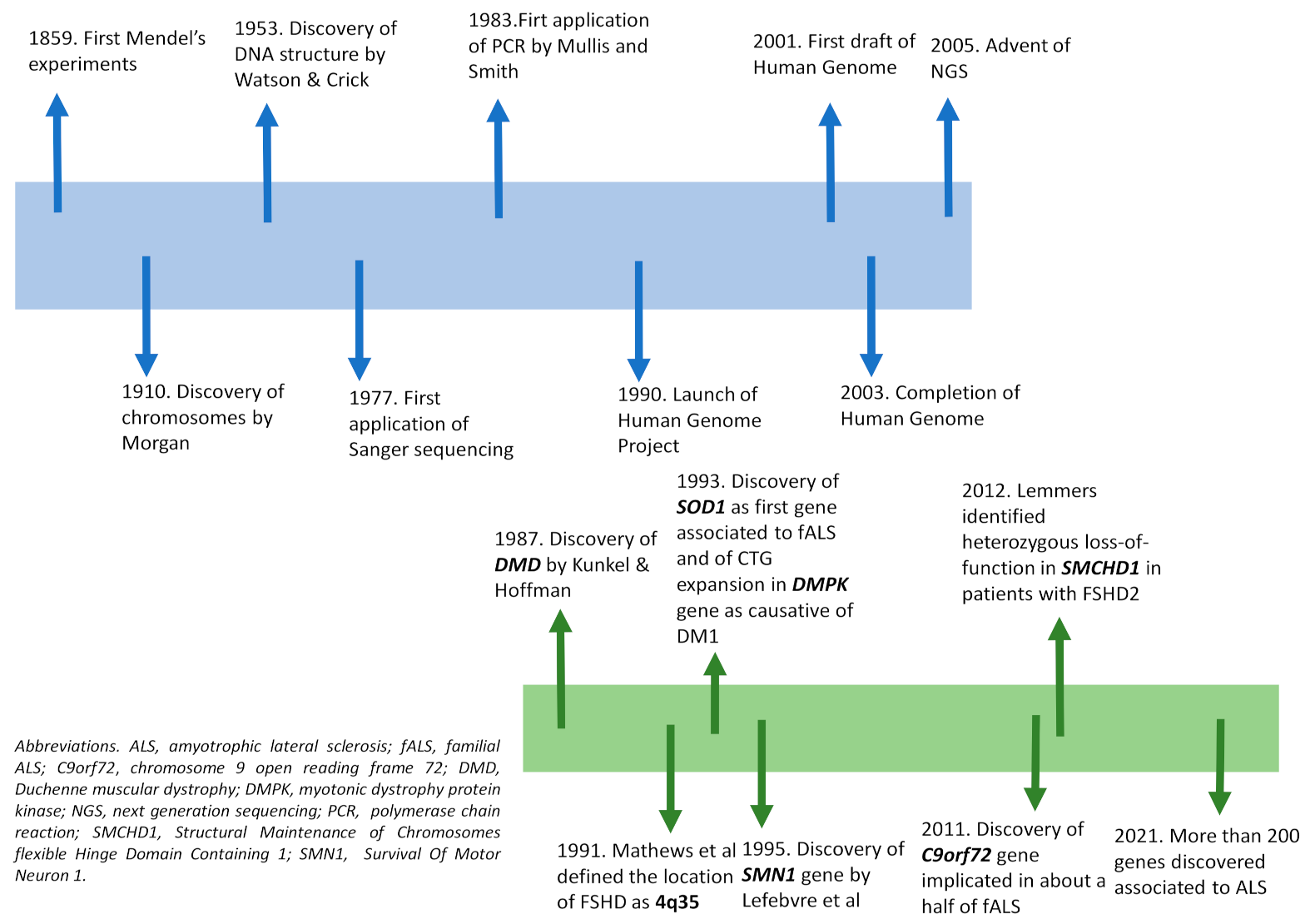{kind=link}
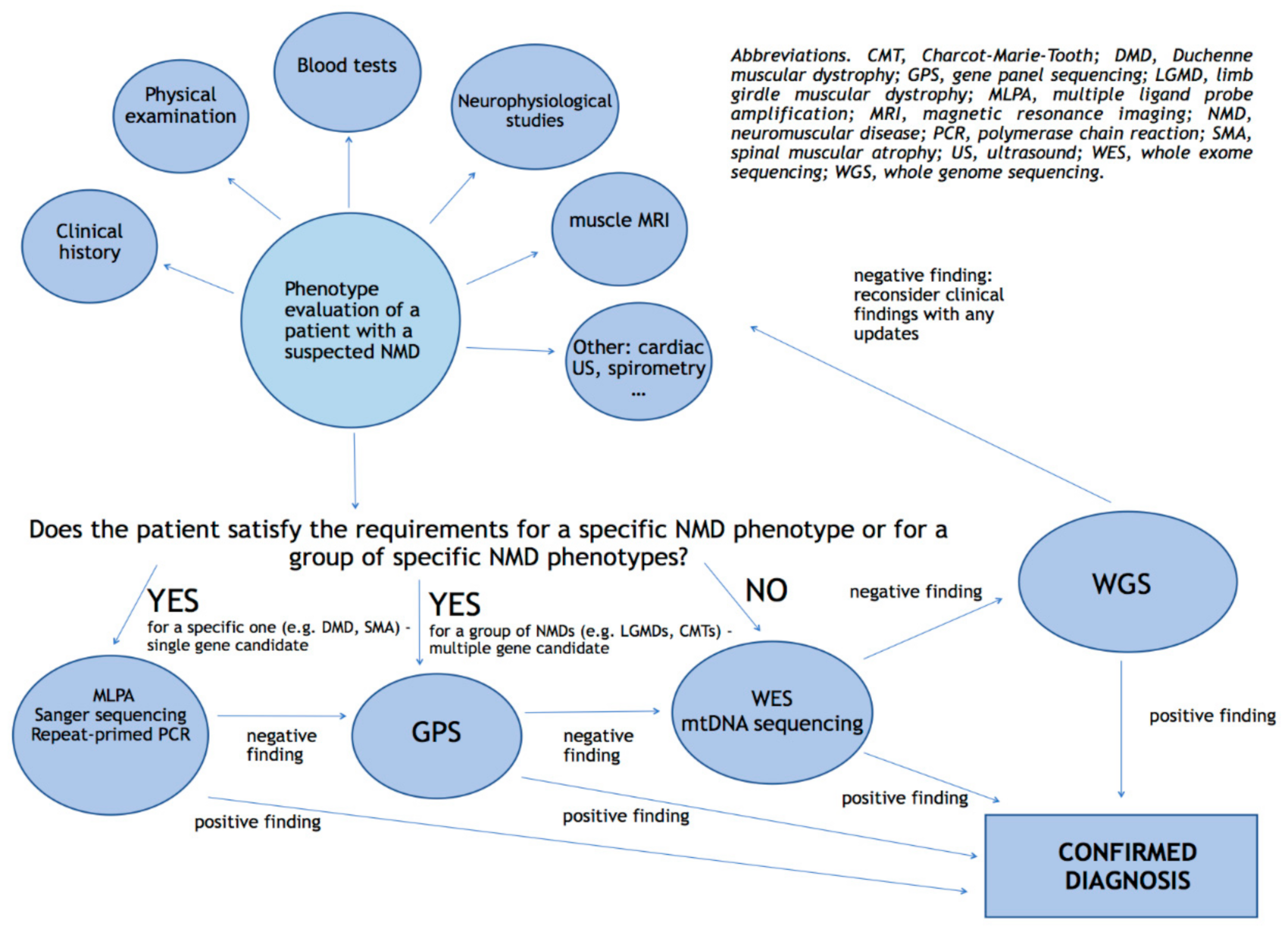{kind=link}
| Neuromuscular Disease | Common Neuromuscular Presentation | Common Extramuscular Presentation | Time-Lag between Onset of Symptoms and Diagnosis |
|---|---|---|---|
| Duchenne muscular dystrophy (DMD) | Very high CK levels Proximal LL weakness Calves hypertrophy | Intellectual disability/autism | 24 months [8] |
| Spinal muscular atrophy (SMA) | Hypotonia and respiratory failure (if birth onset) Proximal muscle weakness and absent DTRs (if adult onset) | _ | 4.7 ± 2.82 months (type 1) 15.6 ± 5.88 months (type 2) 4.34 ± 4.01 years (type 3) [9] |
| Congenital myotonic dystrophy (CDM) | Mixed hypotonia at birth | Intellectual disability Difficulty breathing Swallowing problems Talipes | Few days from birth [10] |
| Myotonic dystrophy type 1 (DM1) | Hand and foot dorsiflexor weakness Hand myotonia Bilteral ptosis Facial weakness | Early-onset cataracts Cardiac arrhythmias Syncope/cardiac arrest Gonadal failure Insulin resistance Excessive daytime sleepiness | 7.3 ± 8.2 years [11] |
| Myotonic dystrophy type 2 (DM2) | High CK Difficulty climbing stairs Muscle pain | Early-onset cataracts Cardiac arrhythmias Insulin resistance Fatiguability | 14.4 ± 12.8 years [11] |
| Facioscapulohumeral muscular dystrophy type 1 and 2 (FSHD1/2) | Proximal weakness in the UL Proximal and distal weakness in the LL Wing scapula Facial weakness | Retinal vasculopathy/Coat syndrome Right bundle branch block High frequency hearing loss Pectus excavatus | Variable, from few years to several years [12] |
| Amyotrophic lateral sclerosis (ALS) | Bulbar onset: dysarthria, dysphagia Spinal onset: weakness in the upper or lower limbs, usually distal | Loss of weight Fatigue Shortness of breath Cognitive impairment | 12 months [13] |
| Main Neuromuscular Sign/Symptom | Possible/Probable Site of Lesion | Differential Diagnosis |
|---|---|---|
| Muscle weakness and stiffness, pseudobulbar signs, ↑↑ DTRs, Babinski and Hoffmann signs, clonus. | UMN | PLS ALS (UMN prevalent) HSP |
| Distal symmetric weakness, distal muscular atrophy, sensory and/or autonomic signs, ↓↓ DTRs, pes cavus, hammertoe deformities, leg atrophy. In general symptoms << signs. | Peripheral nerve | Genetic neuropathy (CMT) |
| Proximal muscle weakness and wasting, ↓↓ or absent DTRs, Gower’s sign, no sensory symptoms. | Skeletal muscle, LMN | Muscular dystrophies SMA type 3 |
| Young age, proximal muscle weakness, facial weakness, diffuse wasting, ↓↓ or absent DTRs, Gower’s sign, bulbar signs, osteoskeletal deformities (pectus excavatus, scoliosis, tendon retractions, congenital hip dysplasia). | Skeletal muscle | CMs |
| Distal muscular weakness, grip myotonia, ↓↓ or absent DTRs, cataract, baldness, ptosis, bulbar signs. | Skeletal muscle | DM1 |
| Proximal muscle weakness, normal or ↑↑ DTR, myotonia, myalgia, cataract | Skeletal muscle | DM2 |
| Limb fasciculations associated with muscle weakness and/or atrophy, ↓↓ or absent DTRs, no sensory symptoms | LMN Peripheral nerve | ALS (LMN prevalent) Kennedy disaease (note that a sensory neuropathy could be also present) Pure motor neuropahy |
| Limb fasciculations associated with muscle weakness and/or atrophy, ↓↓ or absent DTRs, no sensory symptoms, bulbar signs | LMN | ALS (LMN prevalent) Kennedy disaease |
| Mixed LMN and UMN signs in the same myotome (e.g., muscle wasting, ↑↑ DTRs, fasciculations, muscle stiffness), bulbar signs | LMN and UMN | Classic ALS |
| Episodic weakness and/or paralysis | Skeletal muscle (ion channel) | Channelopathies |
| Fluctuating weakness with fatiguability, no sensory symptoms | Neuromuscular junction | Myasthenia gravis |
| Isolated “foot drop” | Peripheral nerve LMN Skeletal muscle | Genetic or acquired neuropathy ALS DM1 FSHD Distal myopathy |
| Isolated “drop head” | LMN Neuromuscular junction Skeletal muscle | ALS Miasthenia gravis Muscular dystrophies Metabolic myopathies |
| Isolated “bulbar signs” | LMN Neuromuscular junction | ALS Myasthenia gravis |
| Hypotonia and/or respiratory failure at birth | LMN Neuromuscular junction Skeletal muscle | SMA type 1 Congenital myasthenia CDM CMDs CMs Congenital myopathies Metabolic myopathy (Pompe disease) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barp, A.; Mosca, L.; Sansone, V.A. Facilitations and Hurdles of Genetic Testing in Neuromuscular Disorders. Diagnostics 2021, 11, 701. https://doi.org/10.3390/diagnostics11040701
Barp A, Mosca L, Sansone VA. Facilitations and Hurdles of Genetic Testing in Neuromuscular Disorders. Diagnostics. 2021; 11(4):701. https://doi.org/10.3390/diagnostics11040701
Chicago/Turabian StyleBarp, Andrea, Lorena Mosca, and Valeria Ada Sansone. 2021. "Facilitations and Hurdles of Genetic Testing in Neuromuscular Disorders" Diagnostics 11, no. 4: 701. https://doi.org/10.3390/diagnostics11040701
APA StyleBarp, A., Mosca, L., & Sansone, V. A. (2021). Facilitations and Hurdles of Genetic Testing in Neuromuscular Disorders. Diagnostics, 11(4), 701. https://doi.org/10.3390/diagnostics11040701