
A New SMAD4 Splice Site Variant in a Three-Generation Italian Family with Juvenile Polyposis Syndrome

 , , , ,
, , , ,  , , , ,
, , , ,  and
and 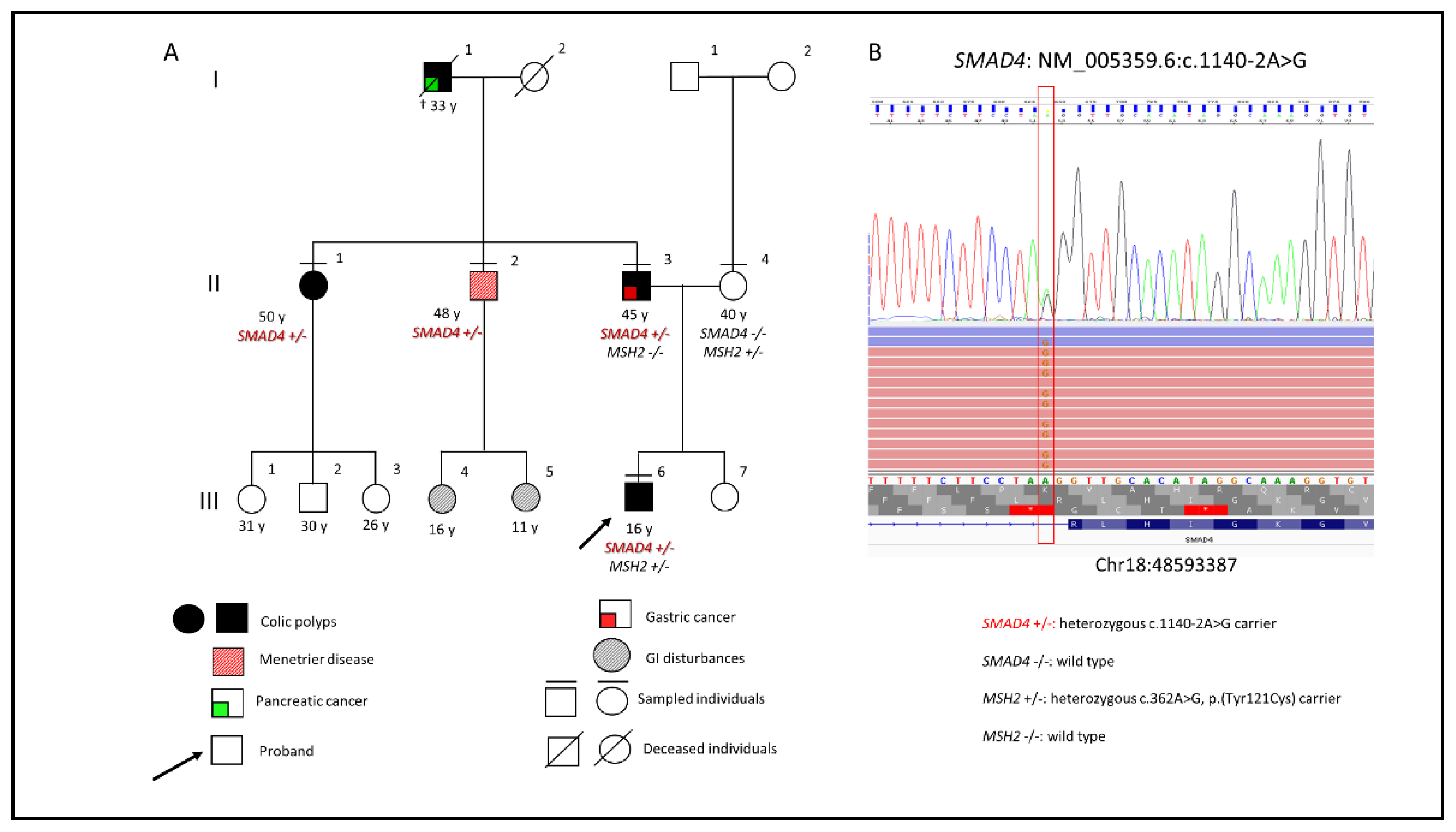{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
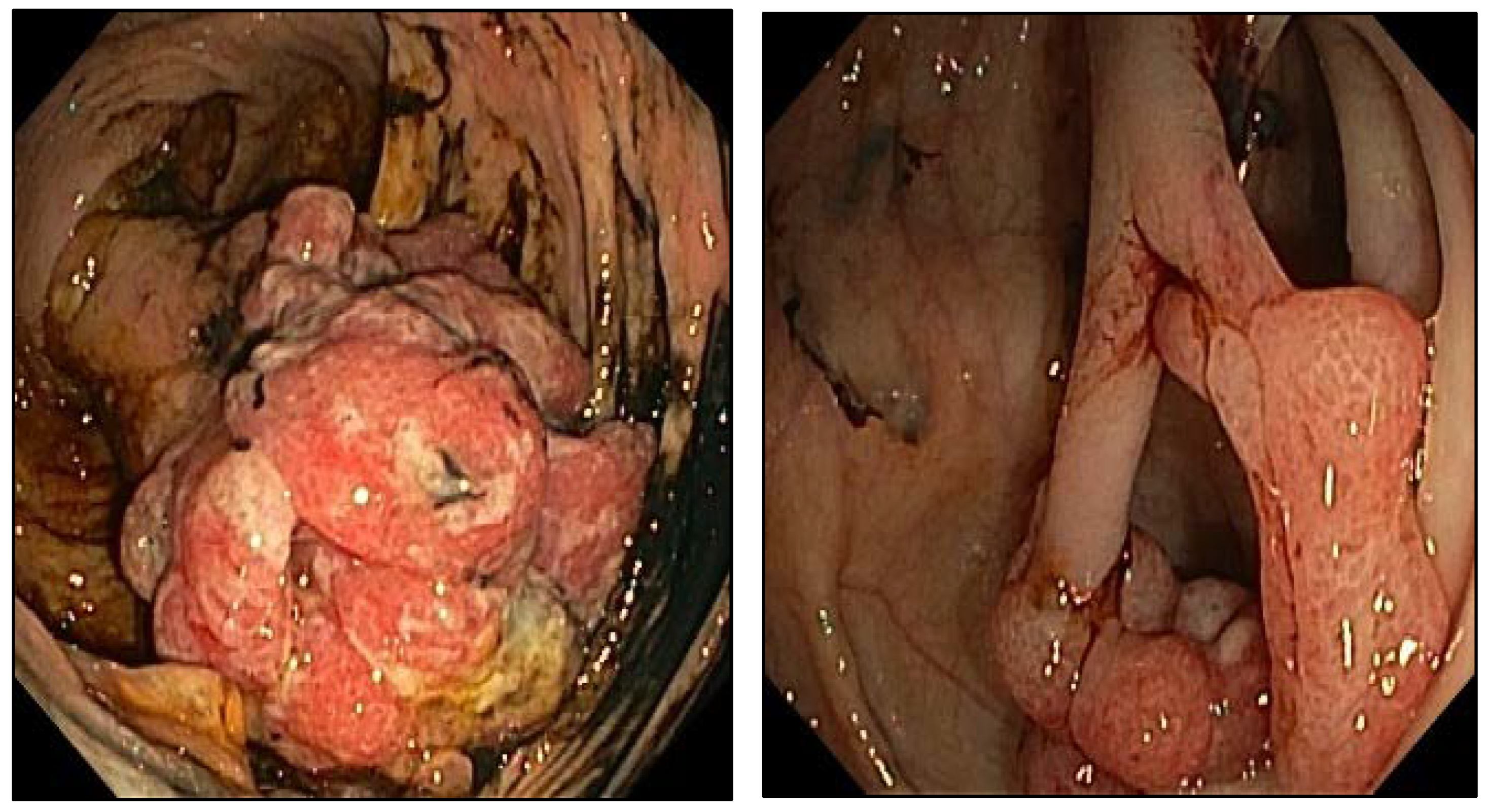
2. Case Presentation
3. Materials and Methods
3.1. NGS Analysis
3.2. RNA Analysis
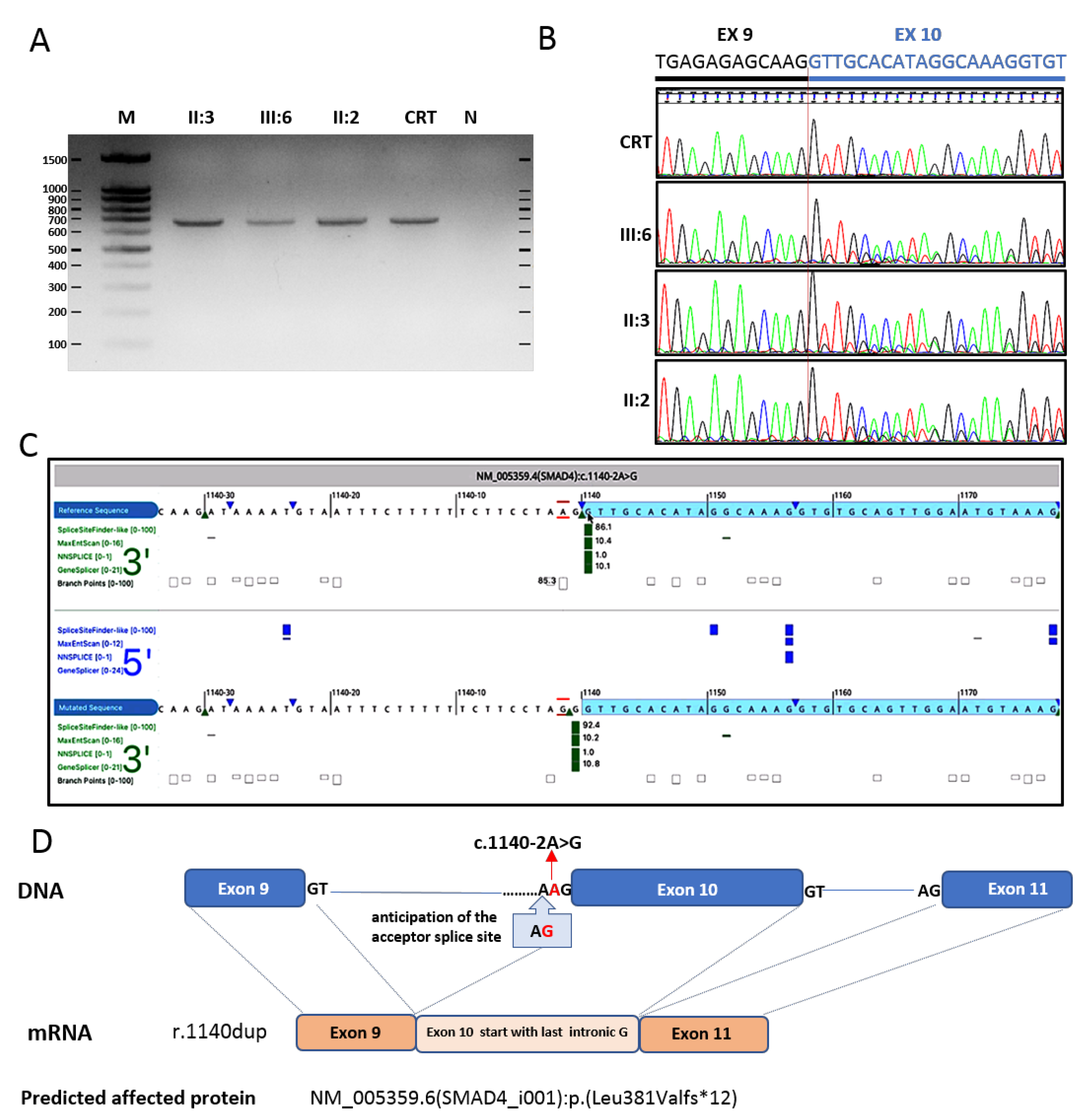
3.3. Validation of SMAD4 Splice Variant
4. Results
5. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Calva, D.; Howe, J.R. Hamartomatous Polyposis Syndromes. Surg. Clin. N. Am. 2008, 88, 779–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigioni, W.F.; Alampi, G.; Martinelli, G.; Piccaluga, A. Atypical juvenile polyposis. Histopathology 1981, 5, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Haidle, J.L.; MacFarland, S.P.; Howe, J.R. Juvenile Polyposis Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993; Available online: http://www.ncbi.nlm.nih.gov/books/NBK1469/ (accessed on 19 April 2022).
- MacFarland, S.P.; Ebrahimzadeh, J.E.; Zelley, K.; Begum, L.; Bass, L.M.; Brand, R.E.; Dudley, B.; Fishman, D.S.; Ganzak, A.; Karloski, E.; et al. Phenotypic Differences in Juvenile Polyposis Syndrome with or Without a Disease-causing SMAD4/BMPR1A Variant. Cancer Prev. Res. 2021, 14, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Kuang, C.; Chen, Y. Tumor-derived C-terminal mutations of Smad4 with decreased DNA binding activity and enhanced intramolecular interaction. Oncogene 2004, 23, 1021–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.-H.; Miyazono, K.; Dijke, P.T. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef]
- Brosens, L.A.; Langeveld, D.; van Hattem, W.A.; Giardiello, F.M.; Offerhaus, G.J. Juvenile polyposis syndrome. World J. Gastroenterol. 2011, 17, 4839–4844. [Google Scholar] [CrossRef]
- O’Malley, M.; LaGuardia, L.; Kalady, M.F.; Parambil, J.; Heald, B.; Eng, C.; Church, J.; Burke, C.A. The Prevalence of Hereditary Hemorrhagic Telangiectasia in Juvenile Polyposis Syndrome. Dis. Colon Rectum 2012, 55, 886–892. [Google Scholar] [CrossRef] [Green Version]
- Blatter, R.; Tschupp, B.; Aretz, S.; Bernstein, I.; Colas, C.; Evans, D.G.; Genuardi, M.; Hes, F.J.; Hüneburg, R.; Järvinen, H.; et al. Disease expression in juvenile polyposis syndrome: A retrospective survey on a cohort of 221 European patients and comparison with a literature-derived cohort of 473 SMAD4/BMPR1A pathogenic variant carriers. Genet. Med. 2020, 22, 1524–1532. [Google Scholar] [CrossRef]
- Sachatello, C.R.; Hahn, I.S.; Carrington, C.B. Juvenile gastrointestinal polyposis in a female infant: Report of a case and review of the literature of a recently recognized syndrome. Surgery 1974, 75, 107–113. [Google Scholar]
- Jia, X.; Burugula, B.B.; Chen, V.; Lemons, R.M.; Jayakody, S.; Maksutova, M.; Kitzman, J.O. Massively parallel functional testing of MSH2 missense variants conferring Lynch syndrome risk. Am. J. Hum. Genet. 2021, 108, 163–175. [Google Scholar] [CrossRef]
- Schwenter, F.; Ratjen, F.; Berk, T.; Gallinger, S.; Gryfe, R.; Gradinger, A.B.; Faughnan, M.E.; Durno, C.A. Juvenile Polyposis Syndrome, SMAD4 Mutations, and Hereditary Hemorrhagic Telangiectasia. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 120–122. [Google Scholar] [CrossRef]
- Pyatt, R.E.; Pilarski, R.; Prior, T.W. Mutation Screening in Juvenile Polyposis Syndrome. J. Mol. Diagn. 2006, 8, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Aretz, S.; Stienen, D.; Uhlhaas, S.; Stolte, M.; Entius, M.M.; Loff, S.; Back, W.; Kaufmann, A.; Keller, K.-M.; Blaas, S.H.; et al. High proportion of large genomic deletions and a genotype phenotype update in 80 unrelated families with juvenile polyposis syndrome. J. Med. Genet. 2007, 44, 702–709. [Google Scholar] [CrossRef] [Green Version]
- Woodford-Richens, K.L.; Rowan, A.J.; Gorman, P.; Halford, S.; Bicknell, D.C.; Wasan, H.S.; Roylance, R.R.; Bodmer, W.F.; Tomlinson, I.P.M. SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 9719–9723. [Google Scholar] [CrossRef] [Green Version]
- Mafficini, A.; Brosens, L.A.A.; Piredda, M.L.; Conti, C.; Mattiolo, P.; Turri, G.; Mastrosimini, M.G.; Cingarlini, S.; Crinò, S.F.; Fassan, M.; et al. Juvenile polyposis diagnosed with an integrated histological, immunohistochemical and molecular approach identifying new SMAD4 pathogenic variants. Fam. Cancer 2022, 1–11. [Google Scholar] [CrossRef]
- Handra-Luca, A.; Condroyer, C.; De Moncuit, C.; Tepper, M.; Fléjou, J.-F.; Thomas, G.; Olschwang, S. Vessels' morphology inSMAD4 andBMPR1A-related juvenile polyposis. Am. J. Med. Genet. Part A 2005, 138A, 113–117. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Aytac, E.; Sulu, B.; Heald, B.; O'Malley, M.; LaGuardia, L.; Remzi, F.H.; Kalady, M.F.; Burke, C.A.; Church, J.M. Genotype-defined cancer risk in juvenile polyposis syndrome. Br. J. Surg. 2015, 102, 114–118. [Google Scholar] [CrossRef]
- Latchford, A.R.; Neale, K.; Phillips, R.K.S.; Clark, S.K. Juvenile Polyposis Syndrome: A study of genotype, phenotype, and long-term outcome. Dis. Colon Rectum 2012, 55, 1038–1043. [Google Scholar] [CrossRef]
- Ishida, H.; Ishibashi, K.; Iwama, T. Malignant tumors associated with juvenile polyposis syndrome in Japan. Surg. Today 2018, 48, 253–263. [Google Scholar] [CrossRef]
- Burmester, J.K.; Bell, L.N.; Cross, D.; Meyer, P.; Yale, S.H.; Meyers, P. A SMAD4 mutation indicative of juvenile polyposis syndrome in a family previously diagnosed with Menetrier's disease. Dig. Liver Dis. 2016, 48, 1255–1259. [Google Scholar] [CrossRef]
- Piepoli, A.; Mazzoccoli, G.; Panza, A.; Tirino, V.; Biscaglia, G.; Gentile, A.; Valvano, M.R.; Clemente, C.; Desiderio, V.; Papaccio, G.; et al. A unifying working hypothesis for juvenile polyposis syndrome and Ménétrier’s disease: Specific localization or concomitant occurrence of a separate entity? Dig. Liver Dis. 2012, 44, 952–956. [Google Scholar] [CrossRef]
- Gallione, C.; Aylsworth, A.S.; Beis, J.; Berk, T.; Bernhardt, B.; Clark, R.D.; Clericuzio, C.; Danesino, C.; Drautz, J.; Fahl, J.; et al. Overlapping spectra of SMAD4 mutations in juvenile polyposis (JP) and JP-HHT syndrome. Am. J. Med. Genet. Part A 2010, 152A, 333–339. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micolonghi, C.; Piane, M.; Germani, A.; Sadeghi, S.; Libi, F.; Savio, C.; Fabiani, M.; Mancini, R.; Ranieri, D.; Pizzuti, A.; et al. A New SMAD4 Splice Site Variant in a Three-Generation Italian Family with Juvenile Polyposis Syndrome. Diagnostics 2022, 12, 2684. https://doi.org/10.3390/diagnostics12112684
Micolonghi C, Piane M, Germani A, Sadeghi S, Libi F, Savio C, Fabiani M, Mancini R, Ranieri D, Pizzuti A, et al. A New SMAD4 Splice Site Variant in a Three-Generation Italian Family with Juvenile Polyposis Syndrome. Diagnostics. 2022; 12(11):2684. https://doi.org/10.3390/diagnostics12112684
Chicago/Turabian StyleMicolonghi, Caterina, Maria Piane, Aldo Germani, Soha Sadeghi, Fabio Libi, Camilla Savio, Marco Fabiani, Rita Mancini, Danilo Ranieri, Antonio Pizzuti, and et al. 2022. "A New SMAD4 Splice Site Variant in a Three-Generation Italian Family with Juvenile Polyposis Syndrome" Diagnostics 12, no. 11: 2684. https://doi.org/10.3390/diagnostics12112684
APA StyleMicolonghi, C., Piane, M., Germani, A., Sadeghi, S., Libi, F., Savio, C., Fabiani, M., Mancini, R., Ranieri, D., Pizzuti, A., Corleto, V. D., Parisi, P., Visco, V., Di Nardo, G., & Petrucci, S. (2022). A New SMAD4 Splice Site Variant in a Three-Generation Italian Family with Juvenile Polyposis Syndrome. Diagnostics, 12(11), 2684. https://doi.org/10.3390/diagnostics12112684