
Microduplication 3p26.3p24.3 and 4q34.3q35.2 Microdeletion Identified in a Patient with Developmental Delay Associated with Brain Malformation


 ,
,  ,
, {kind=link}
{kind=link}
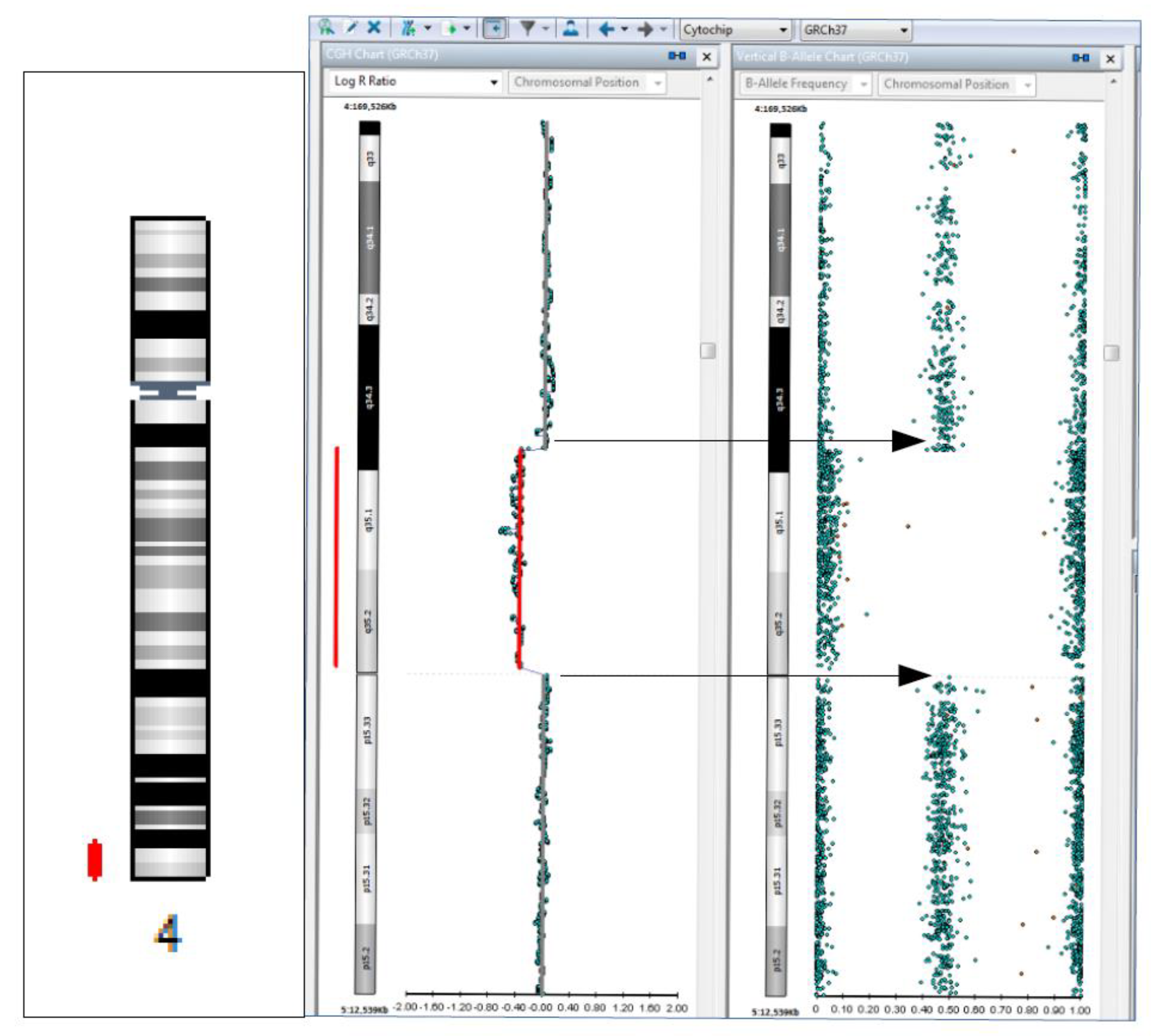
Abstract
:

Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Te Weehi, L.; Maikoo, R.; Mc Cormack, A.; Mazzaschi, R.; Ashton, F.; Zhang, L.; George, A.M.; Love, D.R. Microduplication of 3p26.3 implicated in cognitive development. Case Rep. Genet. 2014, 2014, 295359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Liu, C.; Zhou, B.; Hu, C.; Xu, X. Novel microduplication of CHL1 gene in a patient with autism spectrum disorder: A case report and a brief literature review. Mol. Cytogenet. 2016, 9, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuboyama, M.; Iqbal, M.A. CHL1 deletion is associated with cognitive and language disabilities–Case report and review of literature. Mol. Genet. Genomic. Med. 2021, 9, e1725. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Kuldeep, C.M.; Khare, A.K.; Garg., A.; Mittal, A.; Gupta, L. Terminal 4q deletion syndrome. Indian J. Dermatol. 2012, 57, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Shevell, M.; Ashwal, S.; Donley, D.; Flint, J.; Gingold, M.; Hirtz, D.; Majnemer, A.; Noetzel, M.; Sheth, R.D. Practice parameter: Evaluation of the child with global developmental delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology 2003, 60, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, M.; Hudgins, L. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet. Med. 2010, 12, 742–745. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardos, G.; Gica, N.; Gica, C.; Panaitescu, A.M.; Predescu, M.; Peltecu, G.; Nedelea, F.M. Microduplication 3p26.3p24.3 and 4q34.3q35.2 Microdeletion Identified in a Patient with Developmental Delay Associated with Brain Malformation. Diagnostics 2022, 12, 2887. https://doi.org/10.3390/diagnostics12112887
Cardos G, Gica N, Gica C, Panaitescu AM, Predescu M, Peltecu G, Nedelea FM. Microduplication 3p26.3p24.3 and 4q34.3q35.2 Microdeletion Identified in a Patient with Developmental Delay Associated with Brain Malformation. Diagnostics. 2022; 12(11):2887. https://doi.org/10.3390/diagnostics12112887
Chicago/Turabian StyleCardos, Georgeta, Nicolae Gica, Corina Gica, Anca Maria Panaitescu, Mariana Predescu, Gheorghe Peltecu, and Florina Mihaela Nedelea. 2022. "Microduplication 3p26.3p24.3 and 4q34.3q35.2 Microdeletion Identified in a Patient with Developmental Delay Associated with Brain Malformation" Diagnostics 12, no. 11: 2887. https://doi.org/10.3390/diagnostics12112887
APA StyleCardos, G., Gica, N., Gica, C., Panaitescu, A. M., Predescu, M., Peltecu, G., & Nedelea, F. M. (2022). Microduplication 3p26.3p24.3 and 4q34.3q35.2 Microdeletion Identified in a Patient with Developmental Delay Associated with Brain Malformation. Diagnostics, 12(11), 2887. https://doi.org/10.3390/diagnostics12112887