
Prenatal Diagnosis of Neu–Laxova Syndrome

 and
and 
Abstract
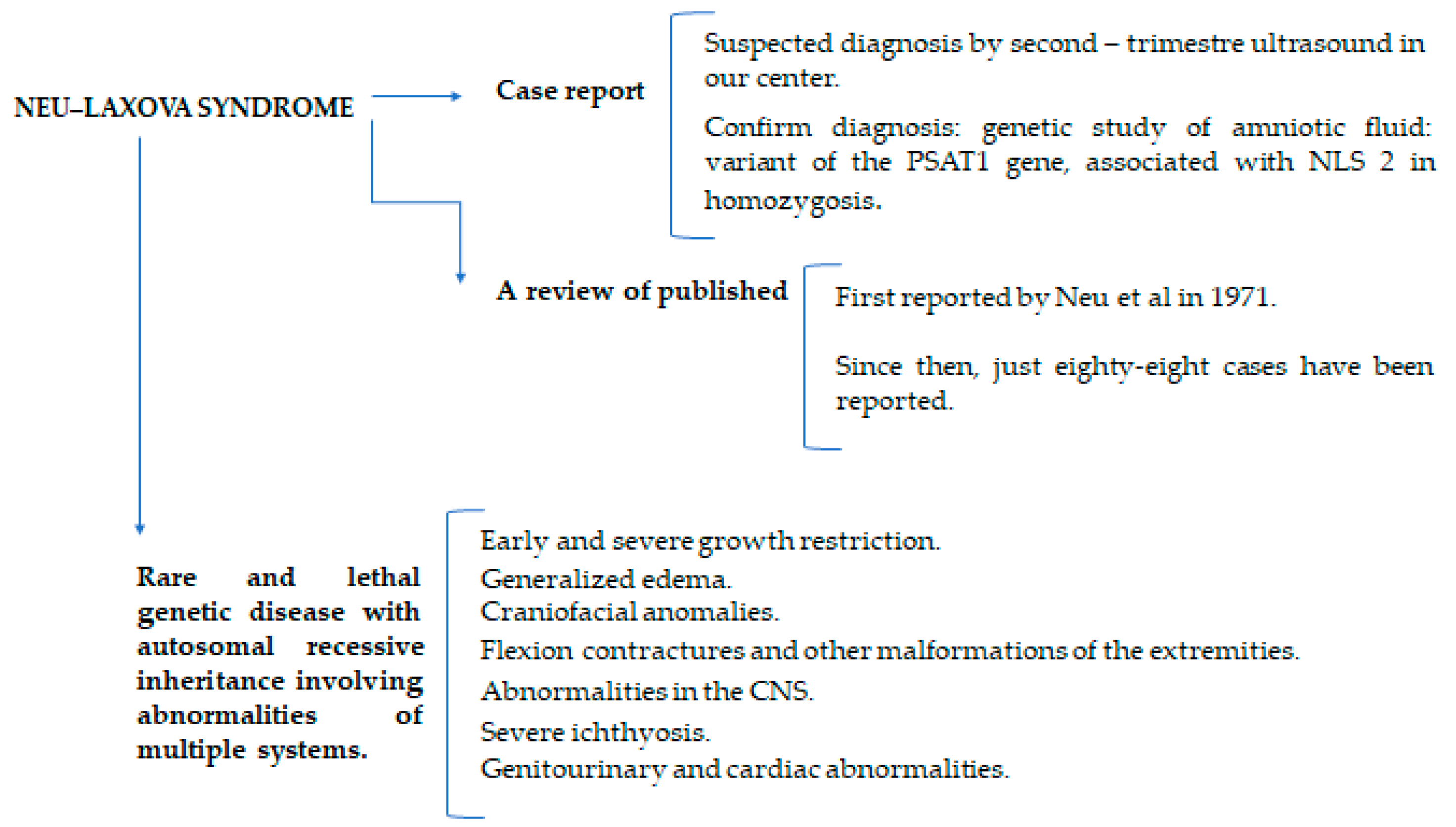
:1. Introduction
2. Materials and Methods
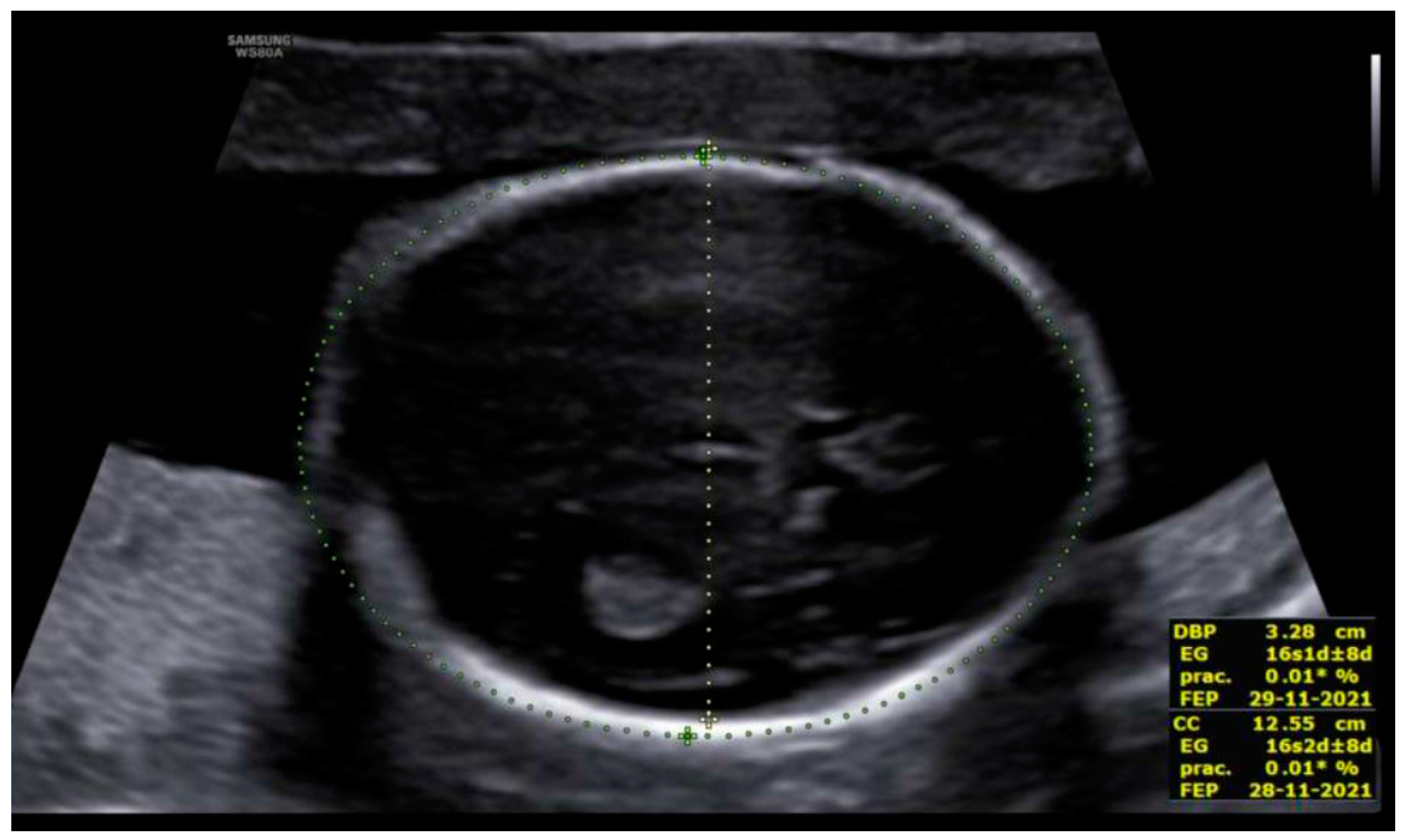
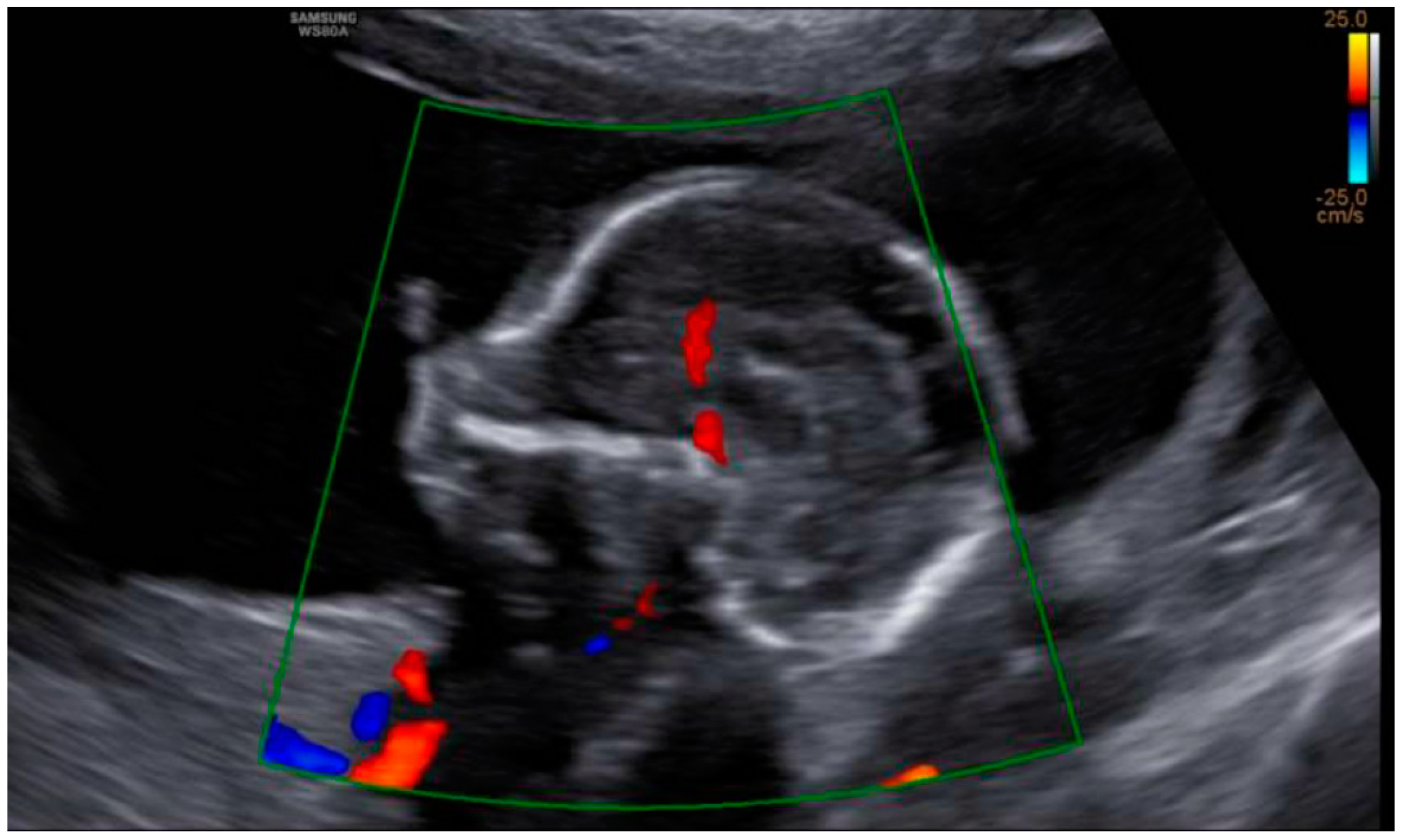
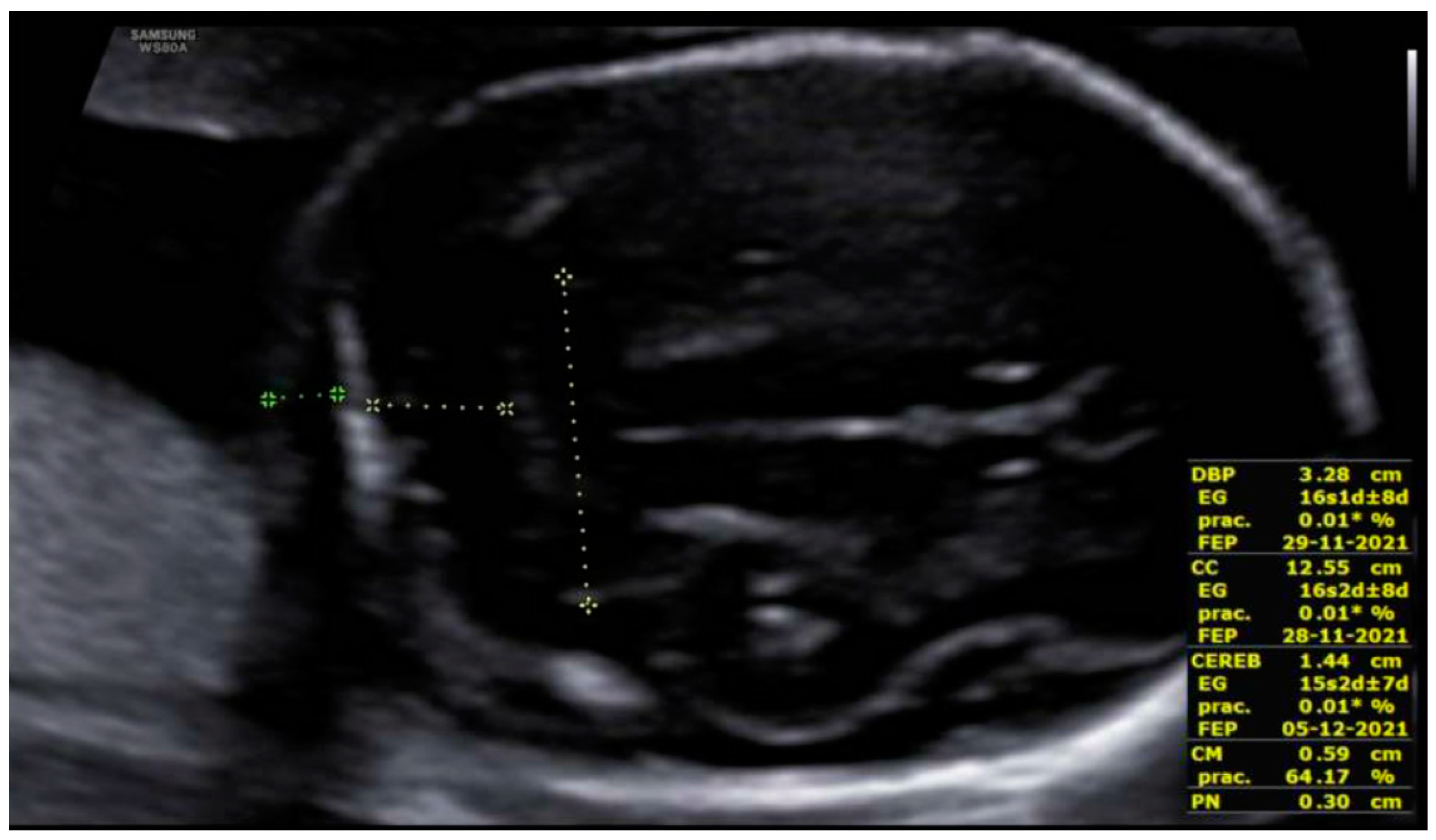
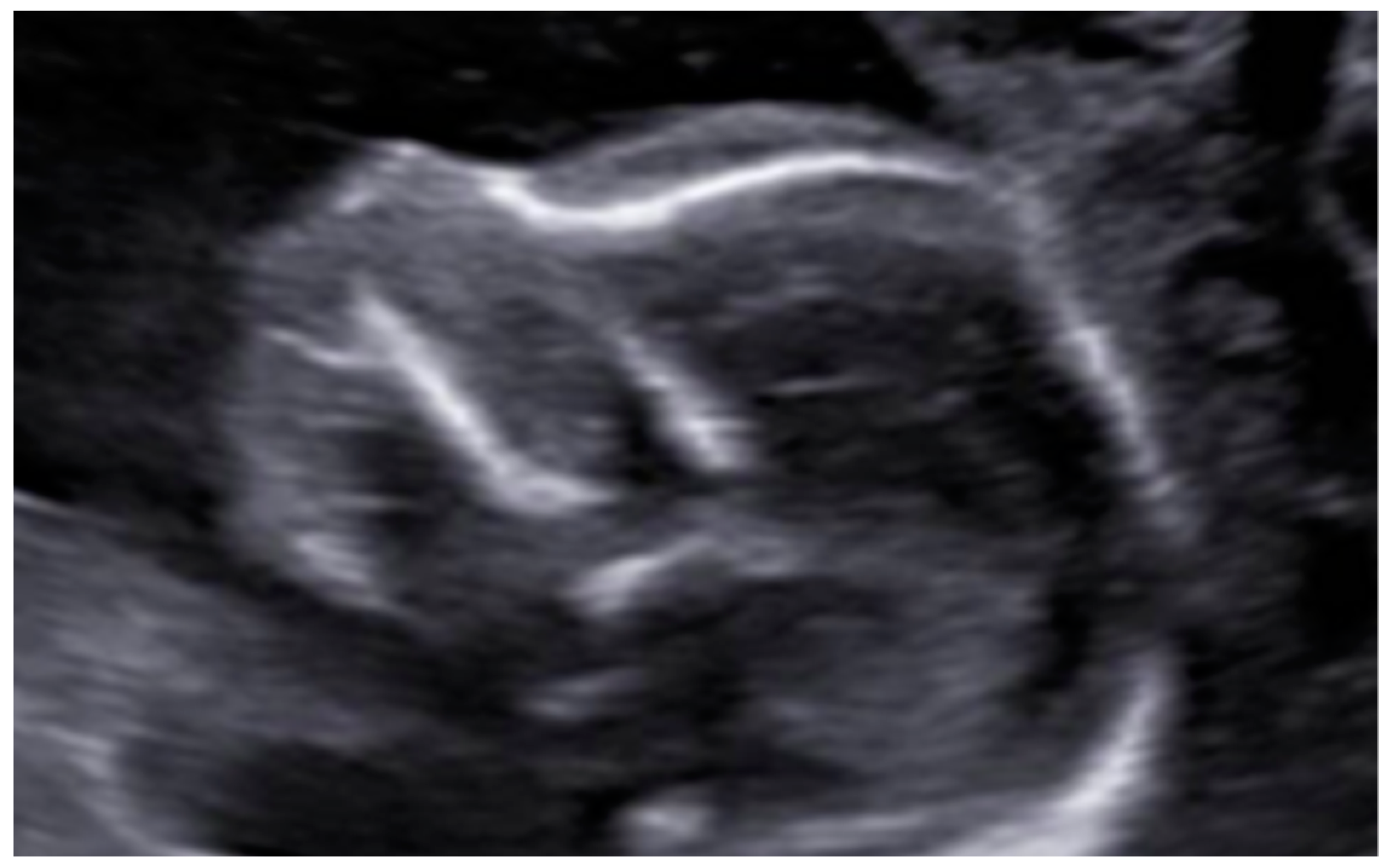
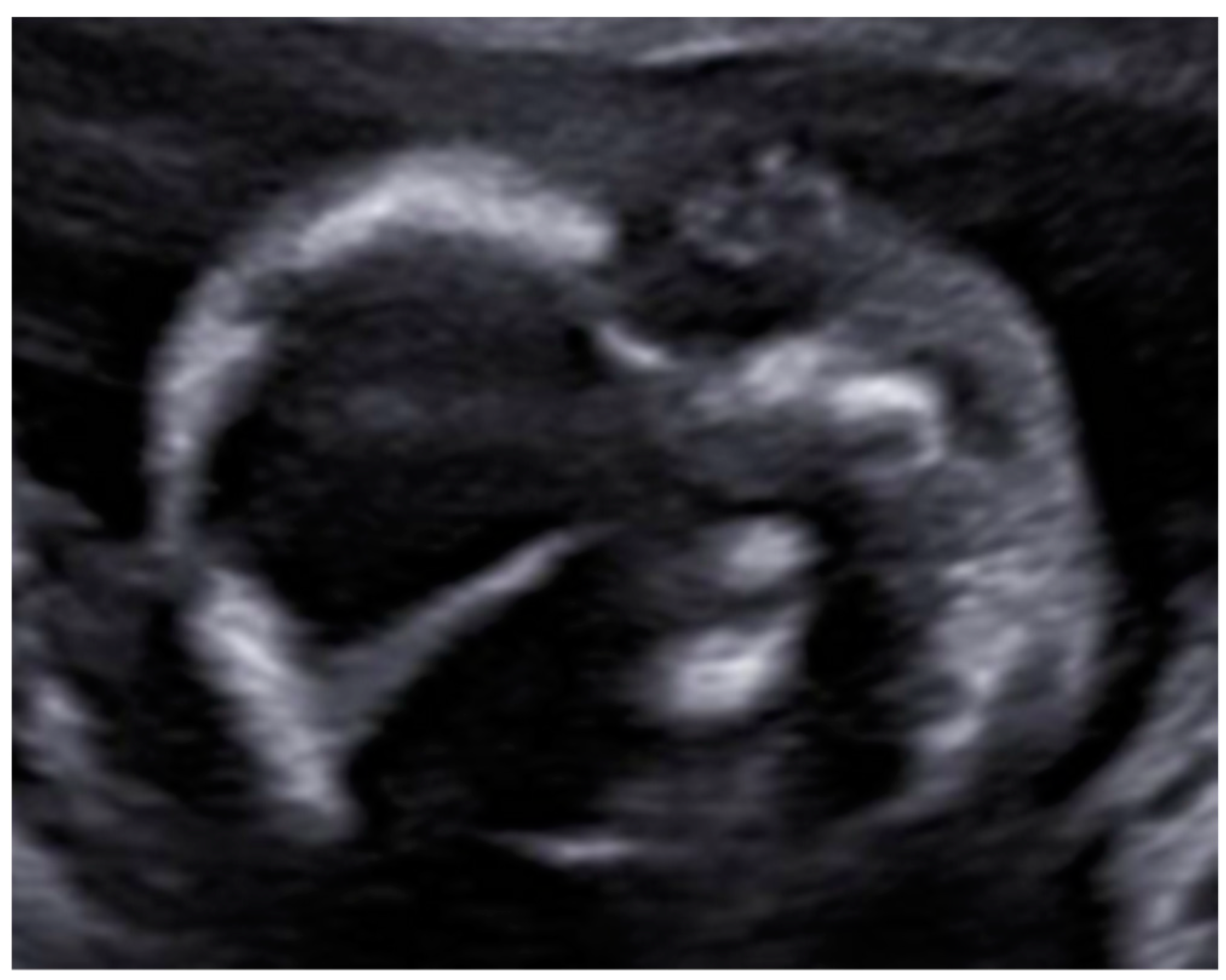
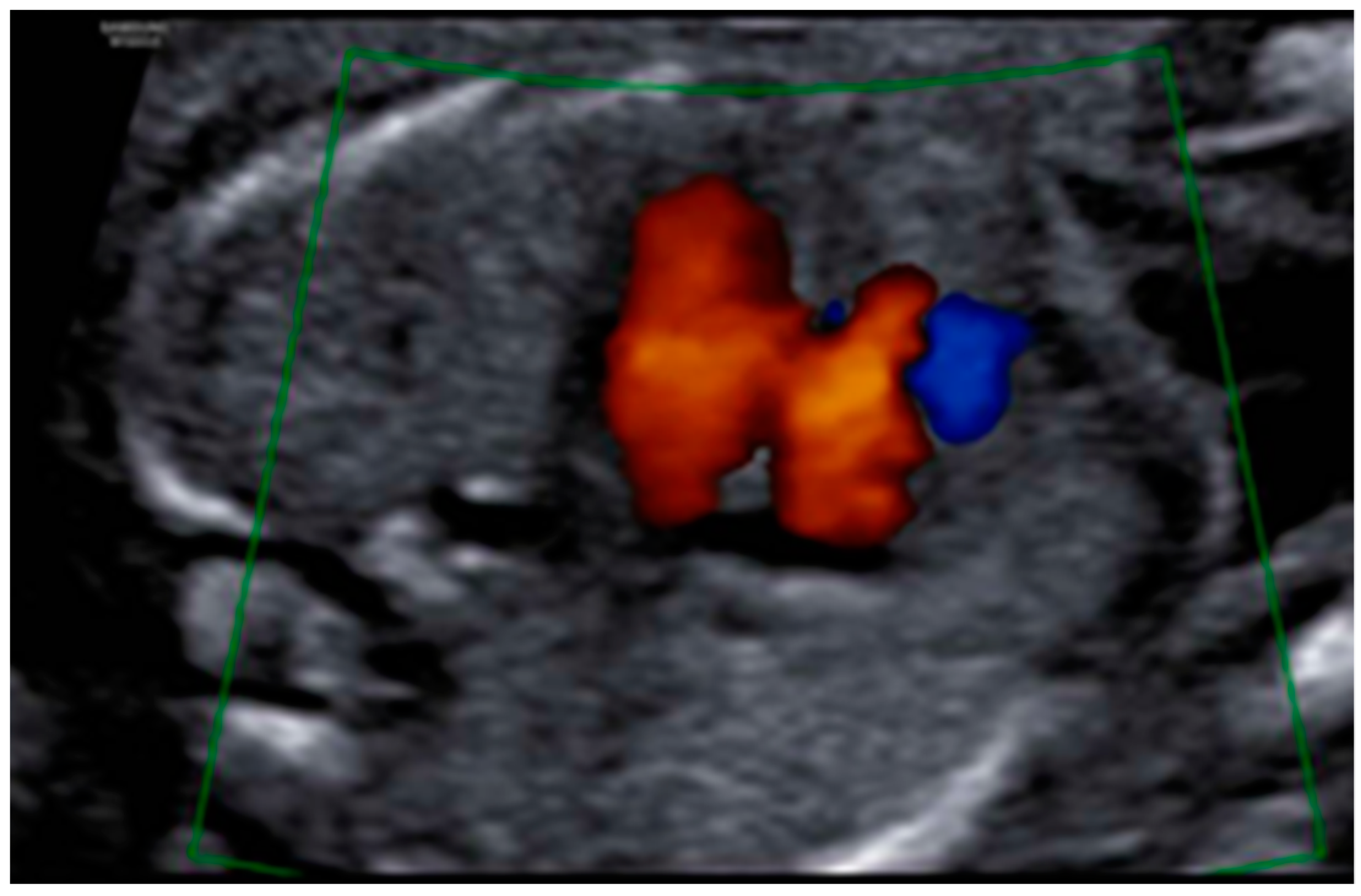
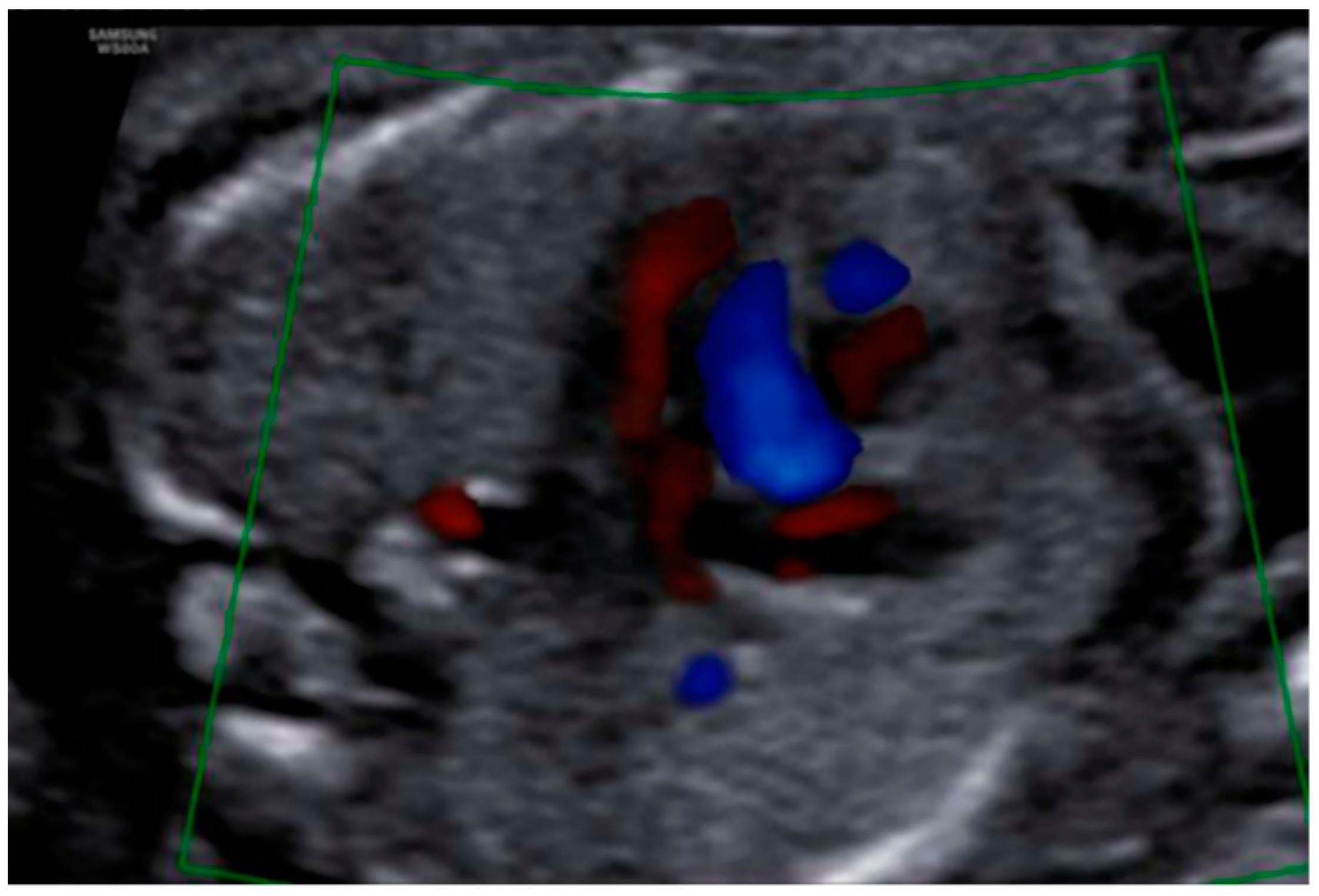
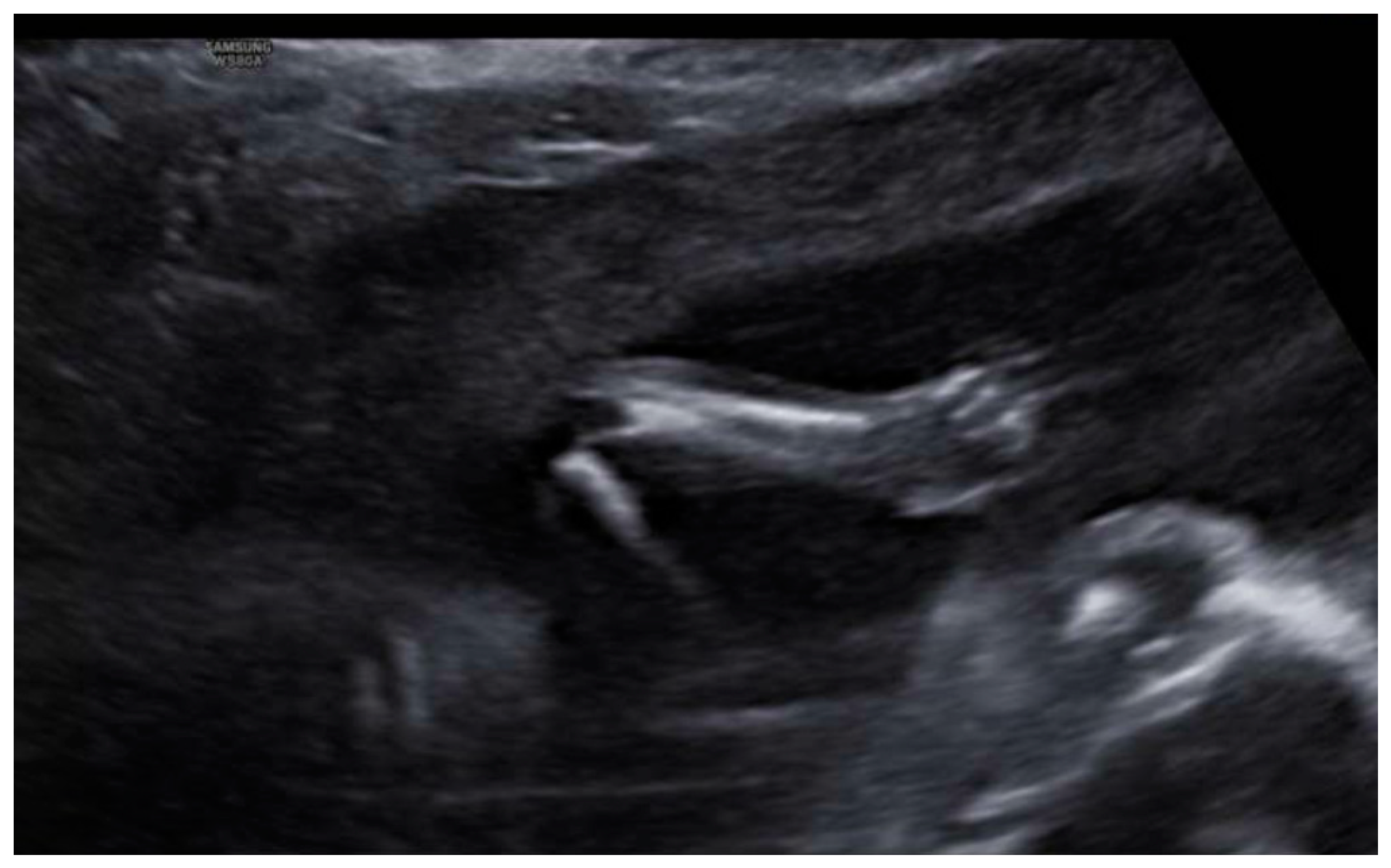
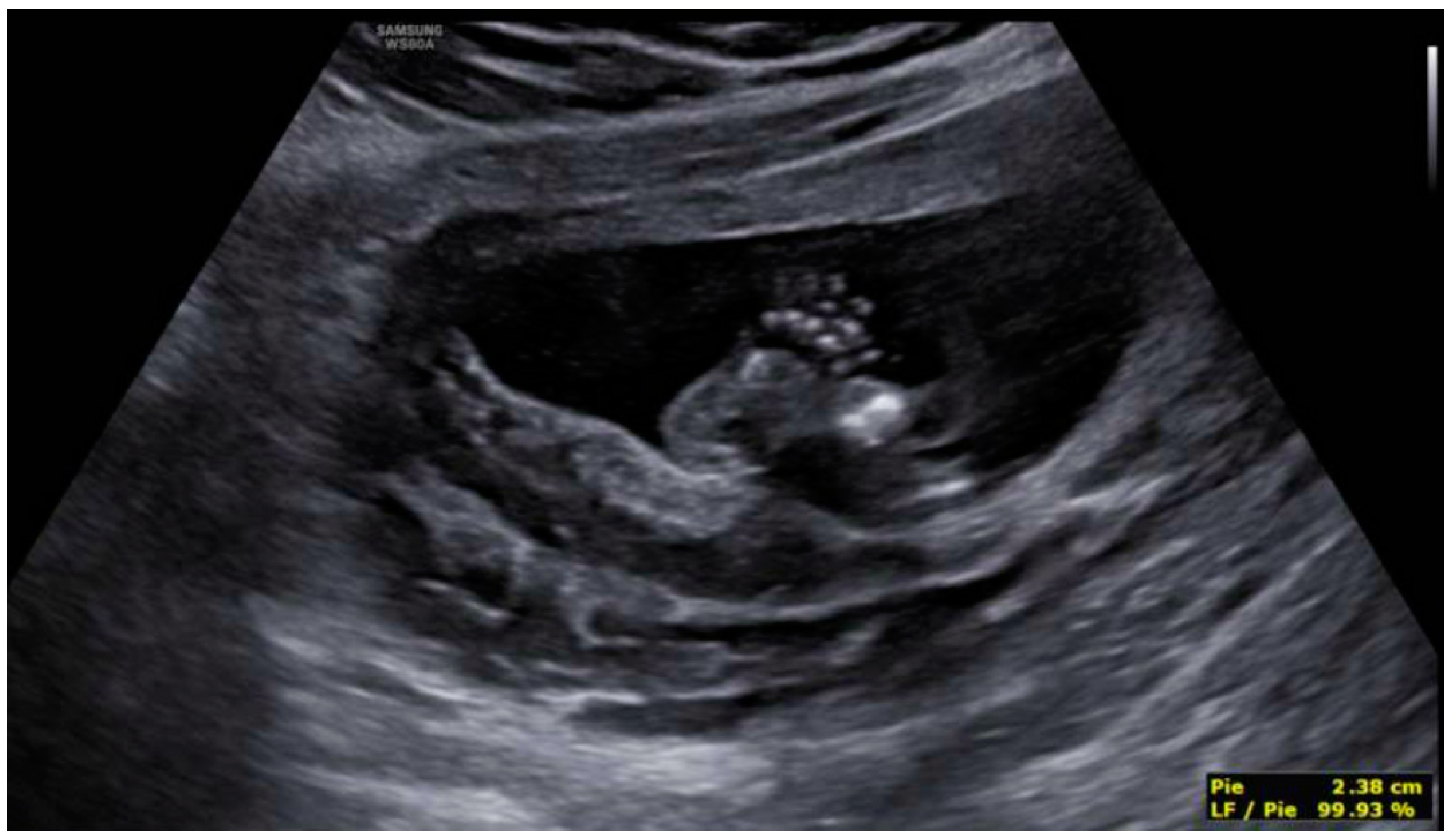
3. Results: Case Presentation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Neu, R.L.; Kajii, T.; Gardner, L.I.; Nagyfy, S.F. A lethal syndrome of microcephaly with multiple congenital anomalies in three siblings. Pediatrics 1971, 47, 610–612. [Google Scholar] [CrossRef] [PubMed]
- Laxova, R.; Ohara, P.T.; Timothy, J.A. A further example of a lethal autosomal recessive condition in sibs. J. Ment. Defic. Res. 1972, 16, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Aslan, H.; Gul, A.; Polat, I.; Mutaf, C.; Agar, M.; Ceylan, Y. Prenatal diagnosis of Neu-Laxova syndrome: A case report. BMC Pregnancy Childbirth 2002, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Darouich, S.; Boujelbene, N.; Kehila, M.; Badis, M.; Reziga, H.; Gaigi, S.; Masmoudi, A. Syndrome de Neu-Laxova: Rapport de trois cas et revue de la littérature. Ann. Pathol. 2016, 36, 235–244. [Google Scholar] [CrossRef]
- Regional Maternal-Child University Hospital of Malaga. Report of the Molecular Genetics Unit, National Molecular Genetics Database, Madrid, February 2021. Available online: http://www.hospitalregionaldemalaga.es/InforCorporativa/UnidadesdeGesti%C3%B3nCl%C3%ADnica/UGCdeLaboratorio/CarteradeServicios.aspx (accessed on 9 May 2022).
- Wood, A.M.; Mottola, A.T.; Rhee, E.H.; Kuller, J.A. Prenatal genetic diagnosis of Neu-Laxova syndrome. J. Obstet. Gynaecol. 2018, 38, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Acuña-Hidalgo, R.; Schanze, D.; Kariminejad, A.; Nordgren, A.; Kariminejad, M.; Conner, P.; Grigelioniene, G.; Nilsson, D.; Nordenskjöld, M.; Wedell, A.; et al. Neu-Laxova syndrome is a heterogeneous metabolic disorder caused by defects in enzymes of the L-serine biosynthesis pathway. Am. J. Hum. Genet. 2014, 95, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Bourque, D.K.; Cloutier, M.; Kernohan, K.D.; Bareke, E.; Grynspan, D.; Michaud, J.; Care4Rare Canada Consortium; Boycott, K. Neu-Laxova syndrome presenting prenatally with increased nuchal translucency and cystic hygroma: The utility of exome sequencing in deciphering the diagnosis. Am. J. Hum. Genet. 2019, 179, 813–816. [Google Scholar] [CrossRef]
- Dwivedi, T.; Gosavi, M. Neu Laxova syndrome. Indian J. Pathol. Microbiol. 2019, 62, 149–152. [Google Scholar] [CrossRef]
- Kahyaoglu, S.; Turgay, I.; Ertas, I.E.; Ceylaner, S.; Danisman, N. Neu-Laxova syndrome, grossly appearing normal on 20 weeks ultrasonographic scan, that manifested late in pregnancy: A case report. Arch. Gynecol. Obstet. 2007, 276, 367–370. [Google Scholar] [CrossRef]
- Driggers, R.W.; Isbister, S.; McShane, C.; Stone, K.; Blakemore, K. Early second trimester prenatal diagnosis of Neu-Laxova syndrome. Prenat. Diagn. 2002, 22, 118–120. [Google Scholar] [CrossRef]
- Ozcan, D.; Derbent, M.; Seçkin, D.; Bikmaz, Y.; Ağildere, M.; De Sandre-Giovannoli, A.; Lévy, N.; Gürakan, B. A collodion baby with facial dysmorphism, limb anomalies, pachygyria and genital hypoplasia: A mild form of Neu-laxova syndrome or a new entity? Ann. Dermatol. 2013, 25, 483–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manar, A.L.; Asma, B. Neu-Laxova syndrome: A new patient with detailed antenatal and post-natal findings. Am. J. Med. Genet. A 2010, 152, 3193–3196. [Google Scholar] [CrossRef] [PubMed]
- Ugras, M.; Kocak, G.; Ozcan, H. Neu-Laxova syndrome: A case report and review of the literature. J. Eur. Acad. Dermatol. Venereol. 2006, 20, 1126–1128. [Google Scholar] [CrossRef] [PubMed]
- Lazjuk, G.I.; Lurie, I.W.; Ostrowskaja, T.I.; Cherstvoy, E.D.; Kirillova, I.A.; Nedzved, M.K. Brief clinical observations: The NeuLaxova syndrome-a distinct entity. Am. J. Med. Genet. 1979, 3, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Mueller, R.F.; Winter, R.M.; Naylor, C.P. Neu-Laxova syndrome: Two further case reports and comments on proposed subclassification. Am. J. Med. Genet. 1983, 16, 645–649. [Google Scholar] [CrossRef]
- Hickey, P.; Piantanida, E.; Lentz-Kapua, S.; Kenner, J. Neu-Laxova syndrome: A case report. Pediatr. Dermatol. 2003, 20, 25–27. [Google Scholar] [CrossRef]
- Karimi-Nejad, M.H.; Khajavi, H.; Gharavi, M.J. Karimi-Nejad, R. NeuLaxova syndrome: Report of a case and comments. Am. J. Med. Genet. 1987, 28, 17–23. [Google Scholar] [CrossRef]
- Meguid, N.A.; Temtamy, S.A. Neu-Laxova syndrome in two Egyptian families. Am. J. Med. Genet. 1991, 41, 30–31. [Google Scholar] [CrossRef]
- Rouzbahani, L. New manifestations in an infant with Neu Laxova syndrome. Am. J. Med. Genet. 1995, 56, 239–240. [Google Scholar] [CrossRef]
- Shved, I.A.; Lazjuk, G.I.; Cherstvoy, E.D. Elaboration of the phenotypic changes of the upper limbs in the Neu-Laxova syndrome. Am. J. Med. Genet. 1985, 20, 1–11. [Google Scholar] [CrossRef]
- Van der Crabben, S.N.; Verhoeven-Duif, N.M.; Brilstra, E.H.; Maldergem, V.L.; Coksun, T.; Rubio-Gozalbo, E. An update on serine deficiency disorders. J. Inherit. Metab. Dis. 2013, 36, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Suranagi, V.; Patil, K.; Bannur, H. Neu-Laxova Syndrome: An Unusual Association with Kyphosis. Turk. Patoloji. Derg. 2018, 34, 259–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Literature (83 Cases) | Case 1 | Case 2 | |
|---|---|---|---|
| Sex | 57% Female; 33% Males | Male | Female |
| Age (weeks) | 12–41 | 21 | 13 |
| Consanguinity | 45% | No | |
| Karyotype | Normal 73% | Normal | |
| Gene | PHGDH, PSAT1 and PSPH | PSAT1 in homozygosis | PSAT1 in heterozygosis |
| IUGR | 87% | Yes | No |
| Hydrops or subcutaneous edema | 73% | Yes | No |
| CNS malformations | |||
| Microcephaly | 85% | Yes | No |
| Hypoplastic cerebellum | 36% | No | No |
| Lissencephaly | 45% | No | No |
| Agenesis/hypoplasia of CC | 36% | Yes | No |
| Ventriculomegaly | 17% | No | No |
| Craniofacial dysmorphism | |||
| Micrognathia | 68% | Yes | No |
| Ocular proptosis | 56% | Yes | No |
| Flattened nose | 79% | Yes | No |
| Flattened forehead | 81% | Yes | No |
| Hypertelorism | 49% | No | No |
| Limb abnormalities | |||
| Arthrogryposis | 80% | Yes | No |
| Syndactyly | 48% | No | No |
| Pulmonary hypoplasia | 39% | No | No |
| Polyhydramnios | 31% | No | No |
| Cardiopathy | 6% | No | No |
| Others | <5% | No | No |
| COFS | Craniofacial malformations, ocular abnormalities, musculoskeletal defects, malformations and progressive degenerative changes of the brain and spinal cord. |
| Walker-Warburg syndrome | Severe congenital oculo-cerebral abnormalities, including lissencephaly and ventriculomegaly. |
| Cerebro-arthrodigital sd | Arthromyodysplasia, dyscephaly, sacral agenesis, and hypoplastic digitis. |
| Pena-Shokeir syndrome type I | Abnormal fetal movement profile, craniofacial malformations, pulmonary hypoplasia, IUGR. |
| Smith-Lemli-Opitz syndrome | Facial anomalies, mental retardation, pre- and postnatal growth disorder and abnormalities in the external genitalia. |
| Miller-Dieker syndrome | It is a variety of lissencephaly, where the brain presents with few or no convolutions. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serrano Olave, A.; López, A.P.; Cruz, M.M.; Rodríguez, S.M.; Narbona Arias, I.; López, J.S.J. Prenatal Diagnosis of Neu–Laxova Syndrome. Diagnostics 2022, 12, 1535. https://doi.org/10.3390/diagnostics12071535
Serrano Olave A, López AP, Cruz MM, Rodríguez SM, Narbona Arias I, López JSJ. Prenatal Diagnosis of Neu–Laxova Syndrome. Diagnostics. 2022; 12(7):1535. https://doi.org/10.3390/diagnostics12071535
Chicago/Turabian StyleSerrano Olave, Adriana, Alba Padín López, María Martín Cruz, Susana Monís Rodríguez, Isidoro Narbona Arias, and Jesús S. Jiménez López. 2022. "Prenatal Diagnosis of Neu–Laxova Syndrome" Diagnostics 12, no. 7: 1535. https://doi.org/10.3390/diagnostics12071535
APA StyleSerrano Olave, A., López, A. P., Cruz, M. M., Rodríguez, S. M., Narbona Arias, I., & López, J. S. J. (2022). Prenatal Diagnosis of Neu–Laxova Syndrome. Diagnostics, 12(7), 1535. https://doi.org/10.3390/diagnostics12071535