
Diagnostics of BAP1-Tumor Predisposition Syndrome by a Multitesting Approach: A Ten-Year-Long Experience

 , , ,
, , , 
 ,
,  , , , ,
, , , , 
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Germline Genome Analyses
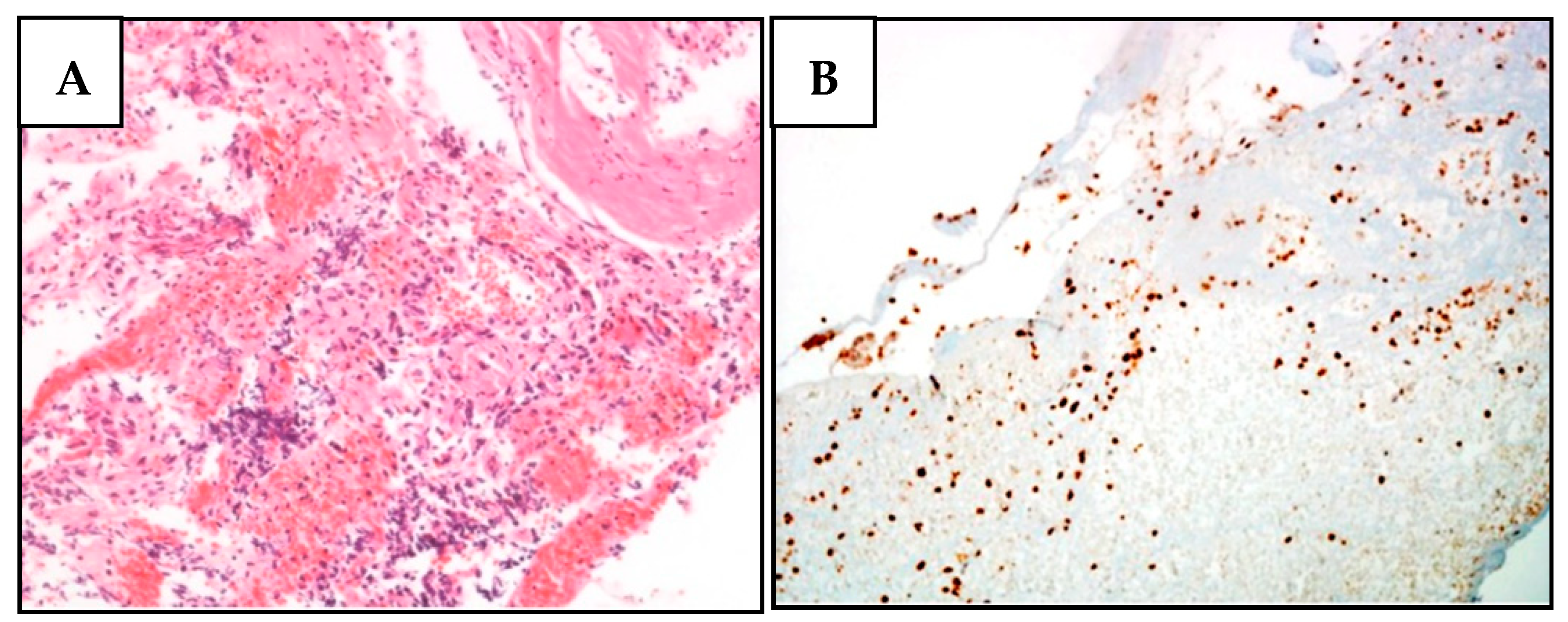
2.3. Immunohistochemical and LOH Analyses
2.4. RT-PCR Analysis of Splice Variant Effects
- -
- forward BAP1-exon 1 (F 5′-ATGAATAAGGGCTGGCTGGAGCT-3′)–reverse BAP1-exon 4 (R 5′-CTGGTGGGCAAAGAACATG-3′) for the HO19.01 patient;
- -
- forward BAP1-exon 5 (F 5′-CCCTGAGTCGCATGAAGGA-3′)–reverse BAP1-exon 7 (R 5′-GTAGACCTTCAGCCCATCCA-3′) for the MM400 patient;
- -
- forward BAP1-exon 8 (F 5′-CGAGGAGTGGACAGACAAG-3′)–reverse BAP1-exon 10 (R 5′-ACTTGTTGCTGGCTGACTTG-3′) for the MM1012 patient.
2.5. Somatic NGS-Targeted Sequencing
2.6. Cascade Testing and Follow-Up
3. Results
3.1. Identification of Families with BAP1-TPDS
3.2. Family A
3.3. Family A1
3.4. Family ID_5
3.5. Family PD-601
3.6. Family ID MPM_HO1901
3.7. Family PD-578
3.8. Family PD-238
4. Discussion
4.1. Should Testing Criteria Be More Stringent?
4.2. The Importance of Detecting Secondary Somatic Mutations of BAP1 in Tumor Tissues
4.3. Which Is the Best Strategy to Detect a Secondary Somatic Mutation?
4.4. Tumor Genome in BAP1-TPDS
4.5. Redefining the Pathogenicity of a Splice Mutation
4.6. Cascade Genetic Testing
4.7. What Can We Say about the TPDS Spectrum?
4.8. New Pathogenic Mutation
4.9. Survival of BAP1 Carriers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bononi, A.; Yang, H.; Giorgi, C.; Patergnani, S.; Pellegrini, L.; Su, M.; Xie, G.; Signorato, V.; Pastorino, S.; Morris, P.; et al. Germline BAP1 Mutations Induce a Warburg Effect. Cell Death Differ. 2017, 24, 1694–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 Mutations Predispose to Malignant Mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, M.; Yang, H. Molecular Pathways: Targeting Mechanisms of Asbestos and Erionite Carcinogenesis in Mesothelioma. Clin. Cancer Res. 2012, 18, 598–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walpole, S.; Hayward, N.K.; Pritchard, A.L.; Johansson, P.A. Microsimulation Model for Evaluating the Cost-Effectiveness of Surveillance in BAP1 Pathogenic Variant Carriers. JCO Clin. Cancer Inform. 2021, 5, 143–154. [Google Scholar] [CrossRef]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and Cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef]
- Pilarski, R.; Rai, K.; Cebulla, C.; Abdel-Rahman, M. BAP1 Tumor Predisposition Syndrome. In GeneReviews®; University of Washington: Seattle, WA, USA, 2022. [Google Scholar]
- Walpole, S.; Pritchard, A.L.; Cebulla, C.M.; Pilarski, R.; Stautberg, M.; Davidorf, F.H.; de la Fouchardière, A.; Cabaret, O.; Golmard, L.; Stoppa-Lyonnet, D.; et al. Comprehensive Study of the Clinical Phenotype of Germline BAP1 Variant-Carrying Families Worldwide. J. Natl. Cancer Inst. 2018, 110, 1328–1341. [Google Scholar] [CrossRef]
- Baumann, F.; Flores, E.; Napolitano, A.; Kanodia, S.; Taioli, E.; Pass, H.; Yang, H.; Carbone, M. Mesothelioma Patients with Germline BAP1 Mutations Have 7-Fold Improved Long-Term Survival. Carcinogenesis 2015, 36, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Carbone, M.; Harbour, J.W.; Brugarolas, J.; Bononi, A.; Pagano, I.; Dey, A.; Krausz, T.; Pass, H.I.; Yang, H.; Gaudino, G. Biological Mechanisms and Clinical Significance of BAP1 Mutations in Human Cancer. Cancer Discov. 2020, 10, 1103–1120. [Google Scholar] [CrossRef]
- Carbone, M.; Arron, S.T.; Beutler, B.; Bononi, A.; Cavenee, W.; Cleaver, J.E.; Croce, C.M.; D’Andrea, A.; Foulkes, W.D.; Gaudino, G.; et al. Tumour Predisposition and Cancer Syndromes as Models to Study Gene-Environment Interactions. Nat. Rev. Cancer 2020, 20, 533–549. [Google Scholar] [CrossRef]
- Rai, K.; Pilarski, R.; Cebulla, C.M.; Abdel-Rahman, M.H. Comprehensive Review of BAP1 Tumor Predisposition Syndrome with Report of Two New Cases. Clin. Genet. 2016, 89, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Carbone, M.; Pass, H.I.; Ak, G.; Alexander, H.R.; Baas, P.; Baumann, F.; Blakely, A.M.; Bueno, R.; Bzura, A.; Cardillo, G.; et al. Medical and Surgical Care of Mesothelioma Patients and Their Relatives Carrying Germline BAP1 Mutations. J. Thorac. Oncol. 2022, 17, 873–889. [Google Scholar] [CrossRef] [PubMed]
- Kobrinski, D.A.; Yang, H.; Kittaneh, M. BAP1: Role in Carcinogenesis and Clinical Implications. Transl. Lung Cancer Res. 2020, 9, S60–S66. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Morrow, B.; Thomas, A.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Gadiraju, M.; Panou, V.; Gao, S.; Mian, I.; et al. Inherited Predisposition to Malignant Mesothelioma and Overall Survival Following Platinum Chemotherapy. Proc. Natl. Acad. Sci. USA 2019, 116, 9008–9013. [Google Scholar] [CrossRef] [Green Version]
- Panou, V.; Gadiraju, M.; Wolin, A.; Weipert, C.M.; Skarda, E.; Husain, A.N.; Patel, J.D.; Rose, B.; Zhang, S.R.; Weatherly, M.; et al. Frequency of Germline Mutations in Cancer Susceptibility Genes in Malignant Mesothelioma. J. Clin. Oncol. 2018, 36, 2863–2871. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, S.; Yoshikawa, Y.; Pass, H.I.; Emi, M.; Nasu, M.; Pagano, I.; Takinishi, Y.; Yamamoto, R.; Minaai, M.; Hashimoto-Tamaoki, T.; et al. A Subset of Mesotheliomas with Improved Survival Occurring in Carriers of BAP1 and Other Germline Mutations. J. Clin. Oncol. 2018, 36, 3485–3494. [Google Scholar] [CrossRef]
- Forde, P.M.; Anagnostou, V.; Sun, Z.; Dahlberg, S.E.; Kindler, H.L.; Niknafs, N.; Purcell, T.; Santana-Davila, R.; Dudek, A.Z.; Borghaei, H.; et al. Durvalumab with Platinum-Pemetrexed for Unresectable Pleural Mesothelioma: Survival, Genomic and Immunologic Analyses from the Phase 2 PrE0505 Trial. Nat. Med. 2021, 27, 1910–1920. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, X.Y.; Li, J.; Zhu, W.W. Prognostic and Clinicopathological Significance of BAP1 Protein Expression in Different Types of Cancer-A Meta-Analysis. Genet. Test. Mol. Biomark. 2018, 22, 115–126. [Google Scholar] [CrossRef]
- Gupta, M.P.; Lane, A.M.; DeAngelis, M.M.; Mayne, K.; Crabtree, M.; Gragoudas, E.S.; Kim, I.K. Clinical Characteristics of Uveal Melanoma in Patients With Germline BAP1 Mutations. JAMA Ophthalmol. 2015, 133, 881–887. [Google Scholar] [CrossRef] [Green Version]
- De la Fouchardière, A.; Cabaret, O.; Savin, L.; Combemale, P.; Schvartz, H.; Penet, C.; Bonadona, V.; Soufir, N.; Bressac-de Paillerets, B. Germline BAP1 Mutations Predispose Also to Multiple Basal Cell Carcinomas. Clin. Genet. 2015, 88, 273–277. [Google Scholar] [CrossRef]
- O’Shea, S.J.; Robles-Espinoza, C.D.; McLellan, L.; Harrigan, J.; Jacq, X.; Hewinson, J.; Iyer, V.; Merchant, W.; Elliott, F.; Harland, M.; et al. A Population-Based Analysis of Germline BAP1 Mutations in Melanoma. Hum. Mol. Genet. 2017, 26, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Shankar, G.M.; Santagata, S. BAP1 Mutations in High-Grade Meningioma: Implications for Patient Care. Neuro. Oncol. 2017, 19, 1447–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, G.M.; Abedalthagafi, M.; Vaubel, R.A.; Merrill, P.H.; Nayyar, N.; Gill, C.M.; Brewster, R.; Bi, W.L.; Agarwalla, P.K.; Thorner, A.R.; et al. Germline and Somatic BAP1 Mutations in High-Grade Rhabdoid Meningiomas. Neuro. Oncol. 2017, 19, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oehl, K.; Vrugt, B.; Wagner, U.; Kirschner, M.B.; Meerang, M.; Weder, W.; Felley-Bosco, E.; Wollscheid, B.; Bankov, K.; Demes, M.C.; et al. Alterations in BAP1 Are Associated with Cisplatin Resistance through Inhibition of Apoptosis in Malignant Pleural Mesothelioma. Clin. Cancer Res. 2021, 27, 2277–2291. [Google Scholar] [CrossRef]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive Genomic Analysis of Malignant Pleural Mesothelioma Identifies Recurrent Mutations, Gene Fusions and Splicing Alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1549–1565. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, Y.; Emi, M.; Hashimoto-Tamaoki, T.; Ohmuraya, M.; Sato, A.; Tsujimura, T.; Hasegawa, S.; Nakano, T.; Nasu, M.; Pastorino, S.; et al. High-Density Array-CGH with Targeted NGS Unmask Multiple Noncontiguous Minute Deletions on Chromosome 3p21 in Mesothelioma. Proc. Natl. Acad. Sci. USA 2016, 113, 13432–13437. [Google Scholar] [CrossRef] [Green Version]
- Righi, L.; Duregon, E.; Vatrano, S.; Izzo, S.; Giorcelli, J.; Rondón-Lagos, M.; Ascoli, V.; Ruffini, E.; Ventura, L.; Volante, M.; et al. BRCA1-Associated Protein 1 (BAP1) Immunohistochemical Expression as a Diagnostic Tool in Malignant Pleural Mesothelioma Classification: A Large Retrospective Study. J. Thorac. Oncol. 2016, 11, 2006–2017. [Google Scholar] [CrossRef] [Green Version]
- Shinozaki-Ushiku, A.; Kohsaka, S.; Kage, H.; Oda, K.; Miyagawa, K.; Nakajima, J.; Aburatani, H.; Mano, H.; Ushiku, T. Genomic Profiling of Multiple Primary Cancers Including Synchronous Lung Adenocarcinoma and Bilateral Malignant Mesotheliomas: Identification of a Novel BAP1 Germline Variant. Pathol. Int. 2020, 70, 775–780. [Google Scholar] [CrossRef]
- Srinivasan, P.; Bandlamudi, C.; Jonsson, P.; Kemel, Y.; Chavan, S.S.; Richards, A.L.; Penson, A.V.; Bielski, C.M.; Fong, C.; Syed, A.; et al. The Context-Specific Role of Germline Pathogenicity in Tumorigenesis. Nat. Genet. 2021, 53, 1577–1585. [Google Scholar] [CrossRef]
- Betti, M.; Aspesi, A.; Biasi, A.; Casalone, E.; Ferrante, D.; Ogliara, P.; Gironi, L.C.; Giorgione, R.; Farinelli, P.; Grosso, F.; et al. CDKN2A and BAP1 Germline Mutations Predispose to Melanoma and Mesothelioma. Cancer Lett. 2016, 378, 120–130. [Google Scholar] [CrossRef]
- Betti, M.; Aspesi, A.; Ferrante, D.; Sculco, M.; Righi, L.; Mirabelli, D.; Napoli, F.; Rondón-Lagos, M.; Casalone, E.; Vignolo Lutati, F.; et al. Sensitivity to Asbestos Is Increased in Patients with Mesothelioma and Pathogenic Germline Variants in BAP1 or Other DNA Repair Genes. Genes Chromosomes Cancer 2018, 57, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A Comprehensive Refinement of the ACMG-AMP Variant Classification Criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Betti, M.; Casalone, E.; Ferrante, D.; Romanelli, A.; Grosso, F.; Guarrera, S.; Righi, L.; Vatrano, S.; Pelosi, G.; Libener, R.; et al. Inference on Germline BAP1 Mutations and Asbestos Exposure from the Analysis of Familial and Sporadic Mesothelioma in a High-Risk Area. Genes Chromosomes Cancer 2015, 54, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Strom, S.P. Current Practices and Guidelines for Clinical Next-Generation Sequencing Oncology Testing. Cancer Biol. Med. 2016, 13, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestergaard, L.K.; Oliveira, D.N.P.; Poulsen, T.S.; Høgdall, C.K.; Høgdall, E.V. OncomineTM Comprehensive Assay v3 vs. OncomineTM Comprehensive Assay Plus. Cancers 2021, 13, 5230. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and Integration of Deleteriousness Prediction Methods for Nonsynonymous SNVs in Whole Exome Sequencing Studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [Green Version]
- Sculco, M.; La Vecchia, M.; Aspesi, A.; Pinton, G.; Clavenna, M.G.; Casalone, E.; Allione, A.; Grosso, F.; Libener, R.; Muzio, A.; et al. Malignant Pleural Mesothelioma: Germline Variants in DNA Repair Genes May Steer Tailored Treatment. Eur. J. Cancer 2022, 163, 44–54. [Google Scholar] [CrossRef]
- Sun, T.; Wang, X.; Wang, M.; Minerowicz, C.; Sanchez, H.; Laskin, W.; Cohen, P.; Zhong, M. Somatic Mutation of BAP1 Can Lead to Expression Loss in Non-Small Cell Lung Carcinoma: Next Generation Sequencing and IHC Analysis in a Large Single Institute Cohort. Int. J. Surg. Pathol. 2021, 30, 512–519. [Google Scholar] [CrossRef]
- Ohar, J.A.; Cheung, M.; Talarchek, J.; Howard, S.E.; Howard, T.D.; Hesdorffer, M.; Peng, H.; Rauscher, F.J.; Testa, J.R. Germline BAP1 Mutational Landscape of Asbestos-Exposed Malignant Mesothelioma Patients with Family History of Cancer. Cancer Res. 2016, 76, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Turunen, J.A.; Markkinen, S.; Wilska, R.; Saarinen, S.; Raivio, V.; Täll, M.; Lehesjoki, A.-E.; Kivelä, T.T. BAP1 Germline Mutations in Finnish Patients with Uveal Melanoma. Ophthalmology 2016, 123, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Boru, G.; Grosel, T.W.; Pilarski, R.; Stautberg, M.; Massengill, J.B.; Jeter, J.; Singh, A.; Marino, M.J.; McElroy, J.P.; Davidorf, F.H.; et al. Germline Large Deletion of BAP1 and Decreased Expression in Non-Tumor Choroid in Uveal Melanoma Patients with High Risk for Inherited Cancer. Genes Chromosomes Cancer 2019, 58, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Njauw, C.-N.J.; Kim, I.; Piris, A.; Gabree, M.; Taylor, M.; Lane, A.M.; DeAngelis, M.M.; Gragoudas, E.; Duncan, L.M.; Tsao, H. Germline BAP1 Inactivation Is Preferentially Associated with Metastatic Ocular Melanoma and Cutaneous-Ocular Melanoma Families. PLoS ONE 2012, 7, e35295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potjer, T.P.; Bollen, S.; Grimbergen, A.J.E.M.; van Doorn, R.; Gruis, N.A.; van Asperen, C.J.; Hes, F.J.; van der Stoep, N. Multigene Panel Sequencing of Established and Candidate Melanoma Susceptibility Genes in a Large Cohort of Dutch Non-CDKN2A/CDK4 Melanoma Families. Int. J. Cancer 2019, 144, 2453–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passiglia, F.; Bironzo, P.; Righi, L.; Listì, A.; Arizio, F.; Novello, S.; Volante, M.; Scagliotti, G.V. A Prospective Phase II Single-Arm Study of Niraparib Plus Dostarlimab in Patients With Advanced Non–small-Cell Lung Cancer and/or Malignant Pleural Mesothelioma, Positive for PD-L1 Expression and Germline or Somatic Mutations in the DNA Repair Genes: Rationale and Study Design. Clin. Lung Cancer 2020, 22, e63–e66. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, Y.; Laitman, Y.; Ben David, M.; Bazak, L.; Lidzbarsky, G.; Salmon, L.B.; Shkedi-Rafid, S.; Barshack, I.; Avivi, C.; Darawshe, M.; et al. Re-Evaluating the Pathogenicity of the c.783+2T>C BAP1 Germline Variant. Hum. Mutat. 2021, 42, 592–599. [Google Scholar] [CrossRef]
- Lin, J.H.; Tang, X.Y.; Boulling, A.; Zou, W.B.; Masson, E.; Fichou, Y.; Raud, L.; Le Tertre, M.; Deng, S.J.; Berlivet, I.; et al. First Estimate of the Scale of Canonical 5’ Splice Site GT>GC Variants Capable of Generating Wild-Type Transcripts. Hum. Mutat. 2019, 40, 1856–1873. [Google Scholar] [CrossRef]
- Cheung, M.; Kadariya, Y.; Talarchek, J.; Pei, J.; Ohar, J.A.; Kayaleh, O.R.; Testa, J.R. Germline BAP1 Mutation in a Family with High Incidence of Multiple Primary Cancers and a Potential Gene-Environment Interaction. Cancer Lett. 2015, 369, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Landry, A.P.; Wang, J.Z.; Nassiri, F.; Patil, V.; Gao, A.; Zadeh, G. BAP1-Deficient Meningioma Presenting with Trabecular Architecture and Cytokeratin Expression: A Report of Two Cases and Review of the Literature. J. Clin. Pathol. 2021. [Google Scholar] [CrossRef]
- Novello, S.; Pinto, C.; Torri, V.; Porcu, L.; Di Maio, M.; Tiseo, M.; Ceresoli, G.; Magnani, C.; Silvestri, S.; Veltri, A.; et al. The Third Italian Consensus Conference for Malignant Pleural Mesothelioma: State of the Art and Recommendations. Crit. Rev. Oncol. Hematol. 2016, 104, 9–20. [Google Scholar] [CrossRef]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and Microsatellite Instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family ID | Patient ID | BAP1 Germline Variant | rs | MAF | Mutation | Localization | Tumor | Reference |
|---|---|---|---|---|---|---|---|---|
| A | A_II-1 § | c.46_47insA | - | - | Insertion | Exon 2 | PeM | [35] |
| A_I-2 | c.46_47insA | - | - | Insertion | Exon 2 | PlM | [35] | |
| A_III-1 | c.46_47insA | - | - | Insertion | Exon 2 | Mucoepidermoid carcinoma | [35] | |
| A_III-2 | c.46_47insA | - | - | Insertion | Exon 2 | Meningioma | This article | |
| A1 | CM | [31] | ||||||
| A1_II-5 § | c.1153C>T p.Arg385* | rs1553645164 | - | Stop gain | Exon 12 | PlM | [31] | |
| BCC | This article | |||||||
| Meningioma | This article | |||||||
| A1_II-2 | c.1153C>T p.Arg385* | rs1553645164 | - | Stop gain | Exon 12 | UM | This article | |
| PlM | This article | |||||||
| A1_III-2 | c.1153C>T p.Arg385* | rs1553645164 | - | Stop gain | Exon 12 | CM | This article | |
| ID_5 | ID_5 III-1 § | c.783+2T>C | rs774730309 | <0.0001 | Donor splice | IVS9 | PlM | [32] |
| PD-601 | MM1012 § | c.783+2T>C | rs774730309 | <0.0001 | Donor splice | IVS9 | CM | This article |
| MPM_HO1901 | PlM | [39] | ||||||
| MPM_HO1901 II-3 § | c.38-1G>T | - | - | Acceptor splice | IVS1 | RCC | [39] | |
| LUAD | [39] | |||||||
| MPM_HO1901 II-2 | c.38-1G>T | - | - | Acceptor splice | IVS1 | PlM | This article | |
| PD-578 | CM | This article | ||||||
| MM981 § | c.605G>A p.Trp202* | - | - | Stop gain | Exon 8 | CM | This article | |
| Prostate cancer | This article | |||||||
| PD-238 | MM400 § | c.376-2A>G | - | Acceptor splice | IVS5 | UM | This article ^ | |
| Bladder cancer | This article ^ | |||||||
| Patient ID | Tumor | Tumor Type | IHC BAP1 | LOH | CNVs | MLPA | FISH | Somatic Variant |
|---|---|---|---|---|---|---|---|---|
| A_II-1 | PeM | NA | NA | NA | NA | NA | NA | / |
| A_I-2 | PlM | FFPE | Not expressed | NA | NA | NA | NA | / |
| A_III-1 | Mucoepidermoid carcinoma | FFPE | Not expressed | Yes | NA | NA | NA | / |
| A_III-2 | Meningioma | FFPE | Not expressed | NA | NA | NA | NA | / |
| A1_II-5 | CM | FFPE | Not expressed | No | NA | NA | NA | / |
| PlM | FFPE | Not expressed | No | NA | NA | NA | / | |
| BCC | FFPE | Not expressed | NA | NA | NA | NA | / | |
| Meningioma | NA | NA | NA | NA | NA | NA | / | |
| A1_II-2 | CM | FFPE | Not expressed | NA | NA | NA | NA | / |
| A1_III-2 | UM | FFPE | Not expressed | NA | NA | NA | NA | / |
| ID_5 III-1 | PlM | FFPE | Not expressed | / | 1.41 (p = 0.006) | NA | 81% deletion ¤ | BAP1 c.783+2T>C (VAF 52.96%) |
| Pleural effusion | / | Yes | / | 50% deletion | 38% deletion § | / | ||
| MM1012 | CM | FFPE | Weak nuclear expression | Yes | NA | NA | NA | / |
| MPM_HO1901 II-3 | PlM | FFPE | Not expressed | NA | 0.96 (p = 0.00004) | NA | NA | BAP1 c.38-1G>T (VAF 89.43%) MSH6 p.Arg1076His (VAF 48%) |
| Pleural effusion | / | Yes | / | 50% deletion | NA | / | ||
| RCC | FFPE | Heterogeneous expression # | NA | / | NA | NA | BAP1 c.38-1G>T (VAF 48.48%) BAP1 p.Trp196* (VAF 2%) MSH6 p.Arg1076His (VAF 48%) | |
| LUAD | FFPE | Not expressed | NA | / | NA | NA | BAP1 c.38-1G>T (VAF 21.20%) BAP1 p.Arg722His (VAF 4.23%) BAP1 p.Arg718Gln (VAF 4.78%) BAP1 p.Arg717Gln (VAF 9%) | |
| MPM_HO1901 II-2 | PlM | FFPE | Not evaluable # | NA | / | NA | NA | BAP1 c.38-1G>T (VAF 53.54%) BAP1 p.Trp196* (VAF 3.36%) MSH6 p.Arg1076His (VAF 25%) |
| MM981 | CM | FFPE | Not expressed | Yes | NA | NA | NA | / |
| CM | FFPE | Not expressed | No | NA | NA | NA | / | |
| Prostate cancer | FFPE | NA | NA | NA | NA | NA | / | |
| MM400 | UM | NA | NA | NA | NA | NA | NA | / |
| Bladder cancer | NA | NA | NA | NA | NA | NA | / |
| Patient ID | Histotype | Age at Diagnosis | Survival |
|---|---|---|---|
| A_II-1 | Epithelioid (Peritoneal) | 63 | 72 months (6 years) * |
| A_I-2 | Epithelioid (Pleural) | 79 | 12 months |
| ID5_II-5 | Epithelioid (Pleural) | 53 | 108 months (9 years) * |
| ID5_II-2 | Epithelioid (Pleural) | 64 | 24 months * |
| ID_5 III-1 | Epithelioid (Pleural) | 52 | 24 months * |
| MPM_HO1901 II-3 | Epithelioid (Pleural) | 58 | 24 months *,° |
| MPM_HO1901 II-2 | Sarcomatoid (Pleural) | 32 | 4 months |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sculco, M.; La Vecchia, M.; Aspesi, A.; Clavenna, M.G.; Salvo, M.; Borgonovi, G.; Pittaro, A.; Witel, G.; Napoli, F.; Listì, A.; et al. Diagnostics of BAP1-Tumor Predisposition Syndrome by a Multitesting Approach: A Ten-Year-Long Experience. Diagnostics 2022, 12, 1710. https://doi.org/10.3390/diagnostics12071710
Sculco M, La Vecchia M, Aspesi A, Clavenna MG, Salvo M, Borgonovi G, Pittaro A, Witel G, Napoli F, Listì A, et al. Diagnostics of BAP1-Tumor Predisposition Syndrome by a Multitesting Approach: A Ten-Year-Long Experience. Diagnostics. 2022; 12(7):1710. https://doi.org/10.3390/diagnostics12071710
Chicago/Turabian StyleSculco, Marika, Marta La Vecchia, Anna Aspesi, Michela Giulia Clavenna, Michela Salvo, Giulia Borgonovi, Alessandra Pittaro, Gianluca Witel, Francesca Napoli, Angela Listì, and et al. 2022. "Diagnostics of BAP1-Tumor Predisposition Syndrome by a Multitesting Approach: A Ten-Year-Long Experience" Diagnostics 12, no. 7: 1710. https://doi.org/10.3390/diagnostics12071710
APA StyleSculco, M., La Vecchia, M., Aspesi, A., Clavenna, M. G., Salvo, M., Borgonovi, G., Pittaro, A., Witel, G., Napoli, F., Listì, A., Grosso, F., Libener, R., Maconi, A., Rena, O., Boldorini, R., Giachino, D., Bironzo, P., Maffè, A., Alì, G., ... Dianzani, I. (2022). Diagnostics of BAP1-Tumor Predisposition Syndrome by a Multitesting Approach: A Ten-Year-Long Experience. Diagnostics, 12(7), 1710. https://doi.org/10.3390/diagnostics12071710