
Integrated Analysis of Tumor Mutation Burden and Immune Infiltrates in Hepatocellular Carcinoma

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Download
2.2. Data Preprocessing and Differentially Expressed Gene (DEG) Screening
2.3. GSEA and GSVA Analysis
2.4. Verification of Differential Expression of NCBP2
2.5. Prognostic Analysis
2.6. Expression Verification of NCBP2 in Cells and Tissues
2.7. Correlation Analysis between NCBP2 and Immunity
3. Results
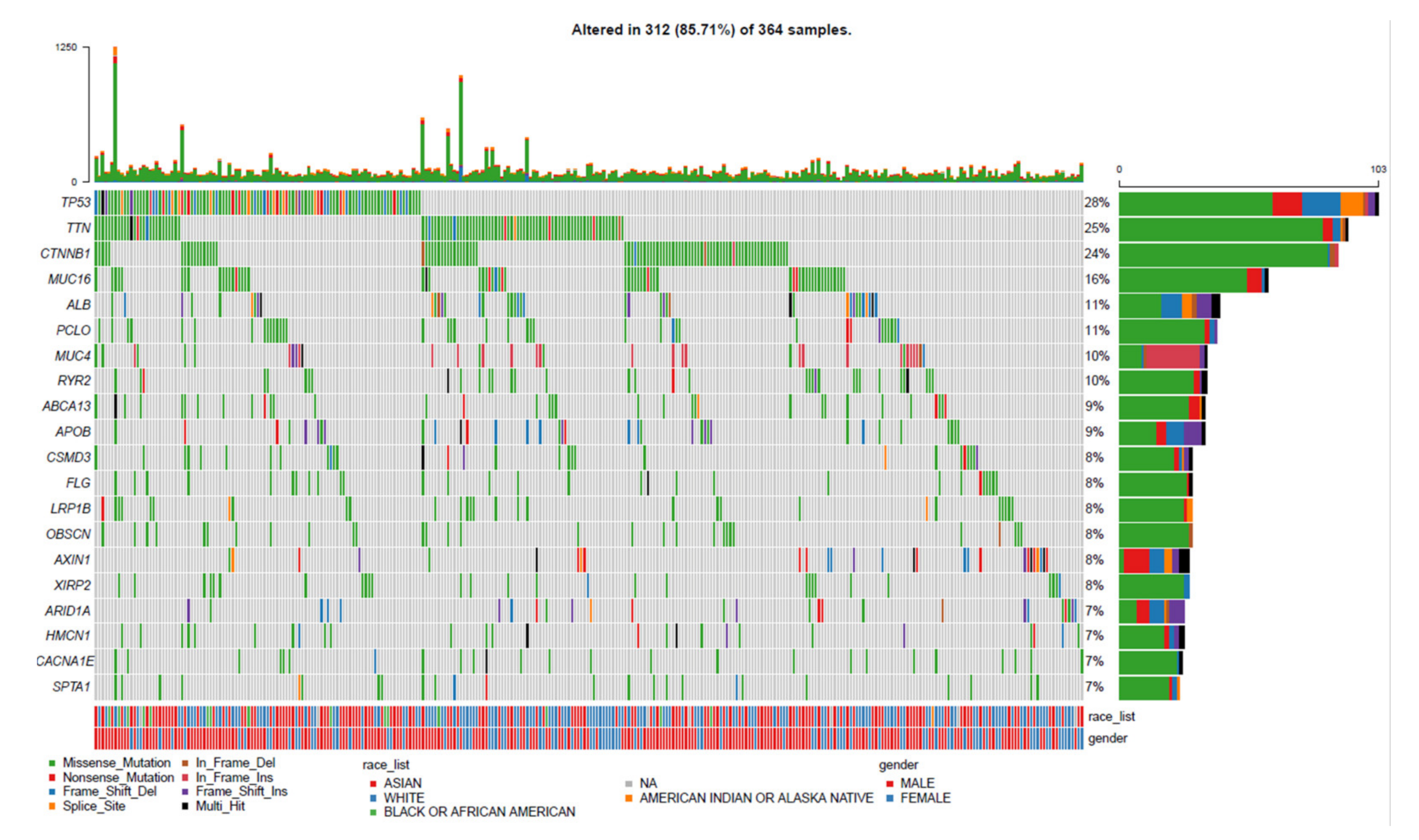
3.1. Landscape of Gene Mutation Files in LIHC
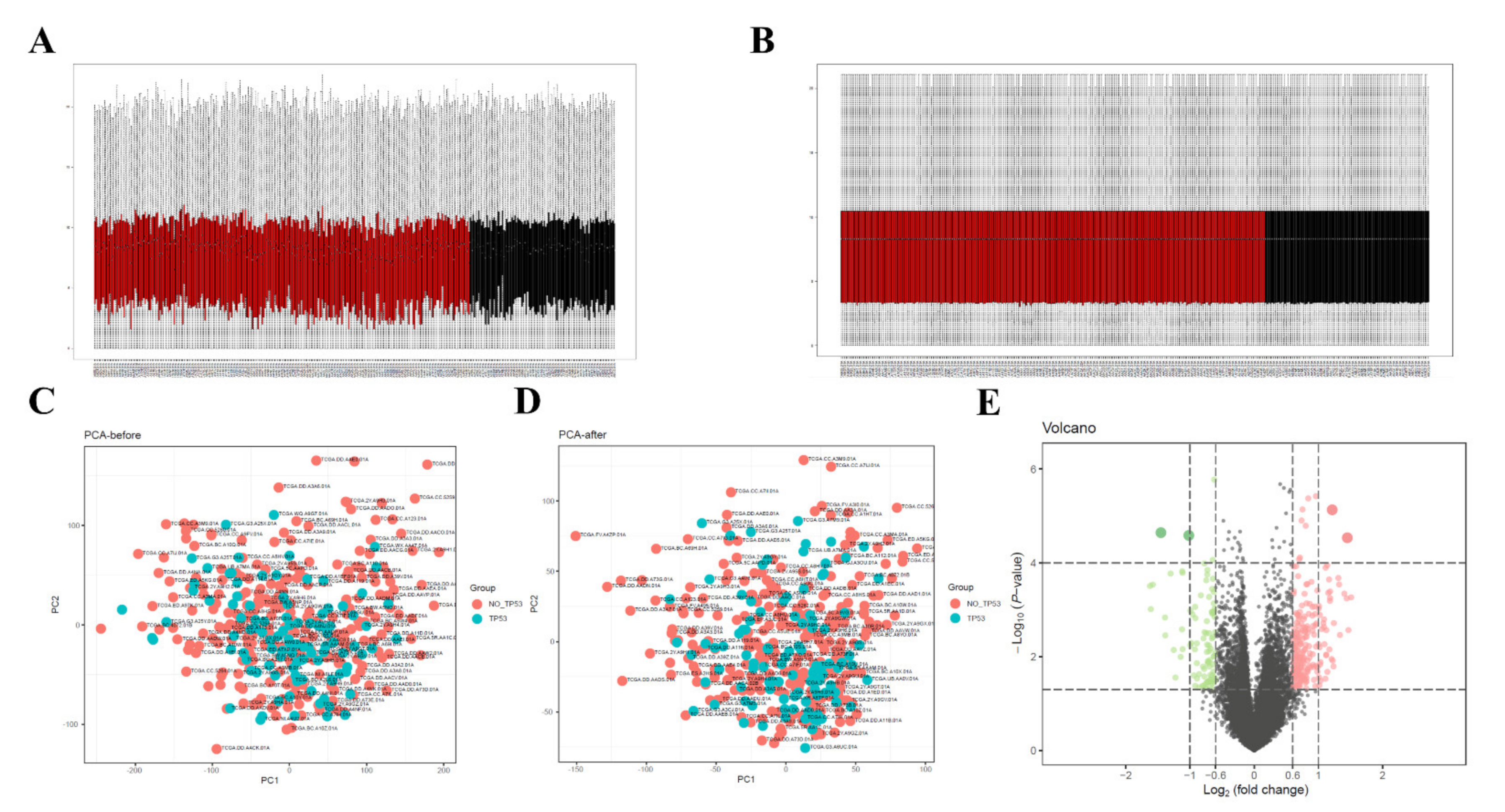
3.2. Data Preprocessing and Screening of DEGs
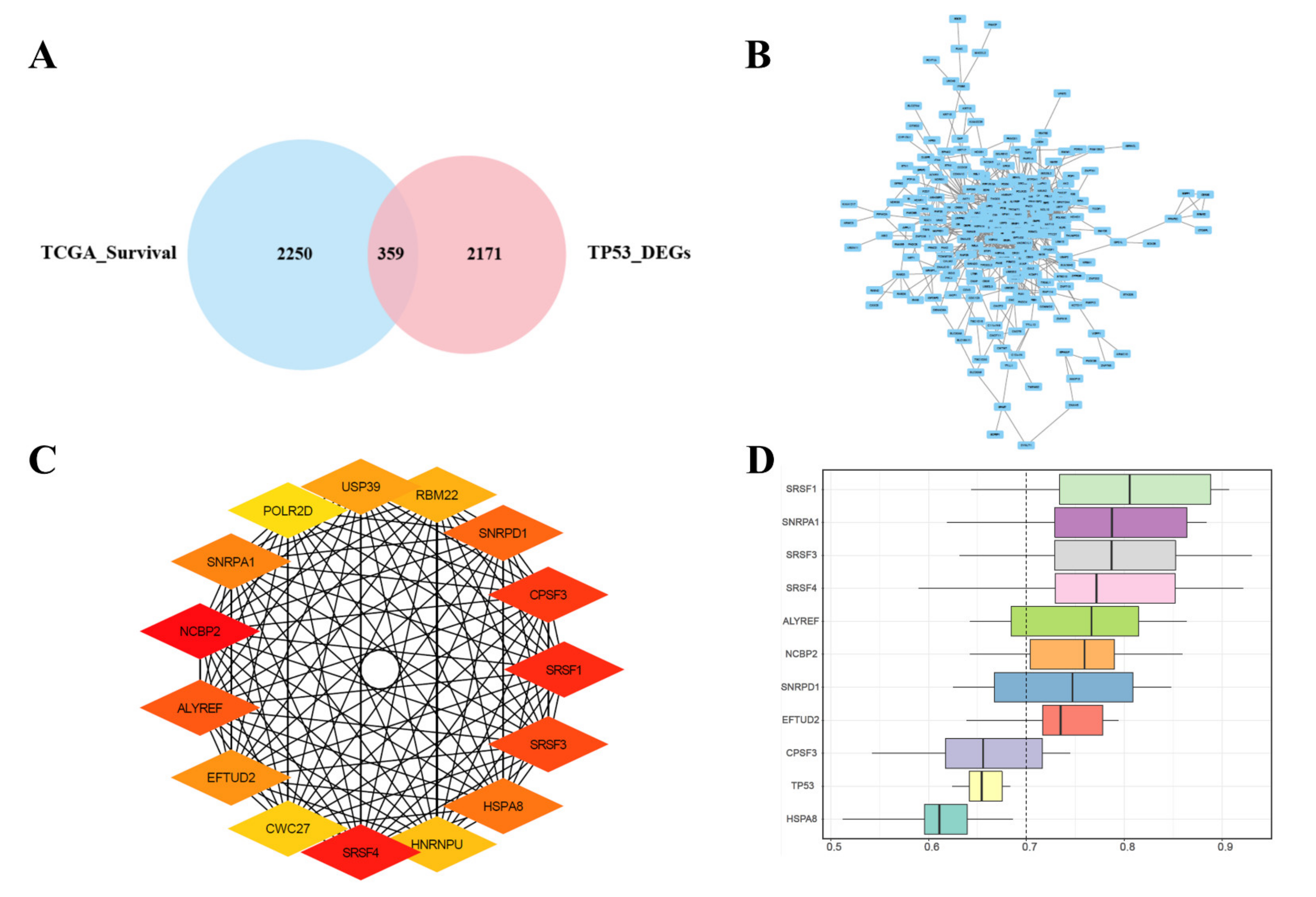
3.3. Joint Screening of Genes, PPI Network Construction, Hub Genes Screening, and Similarities
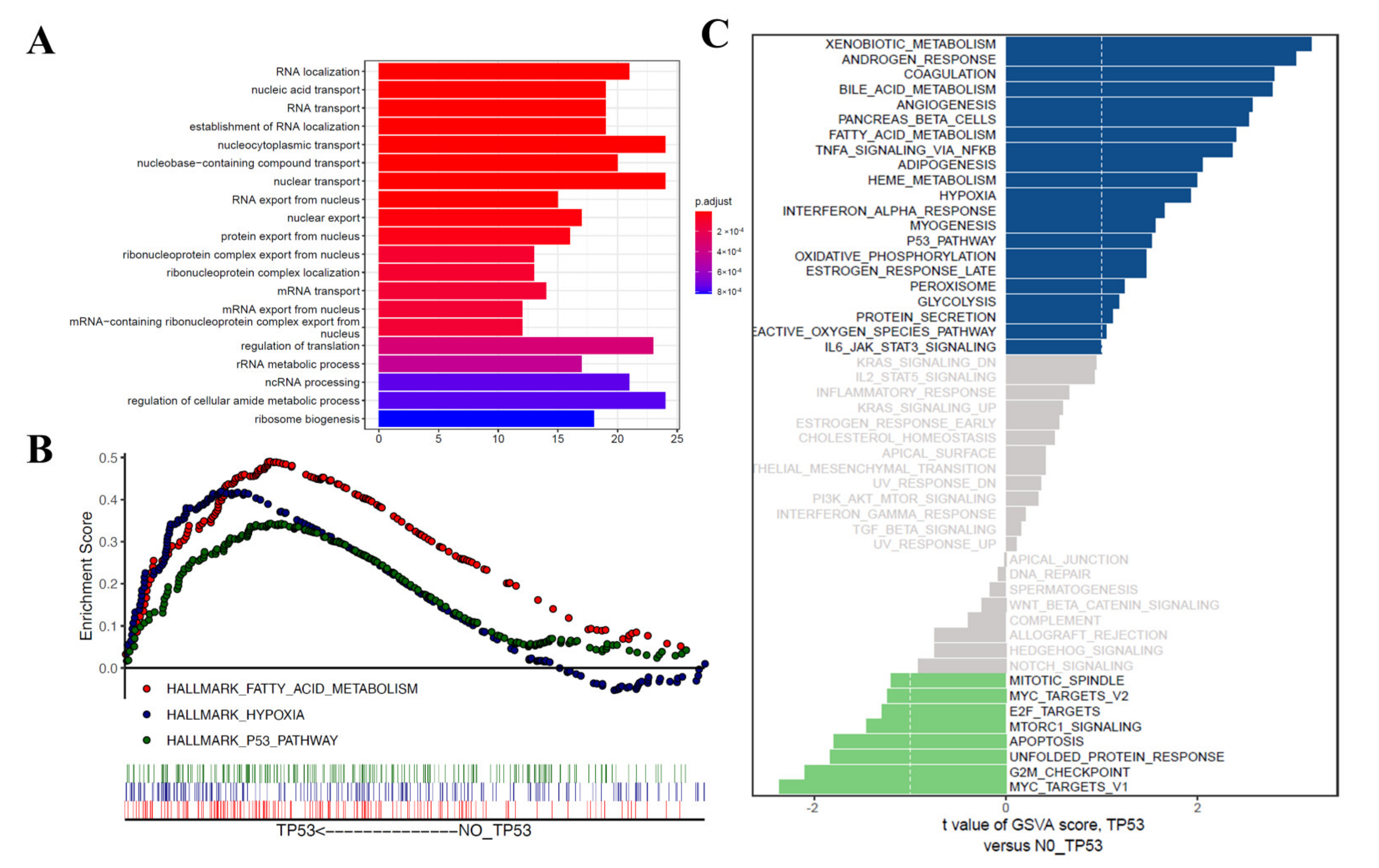
3.4. Functional Correlation Analysis
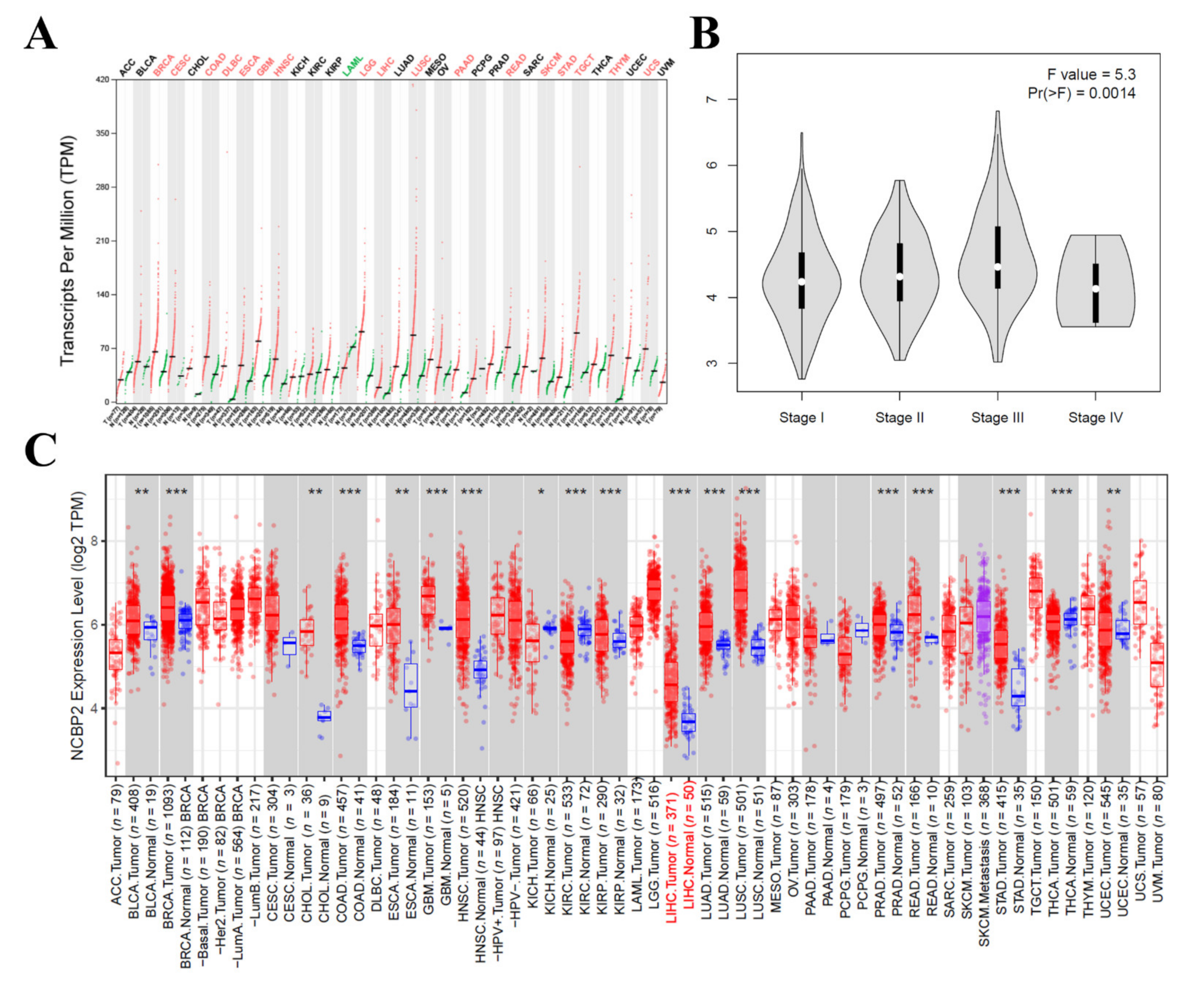
3.5. The mRNA Expression Level of NCBP2 in Hepatocellular Carcinoma
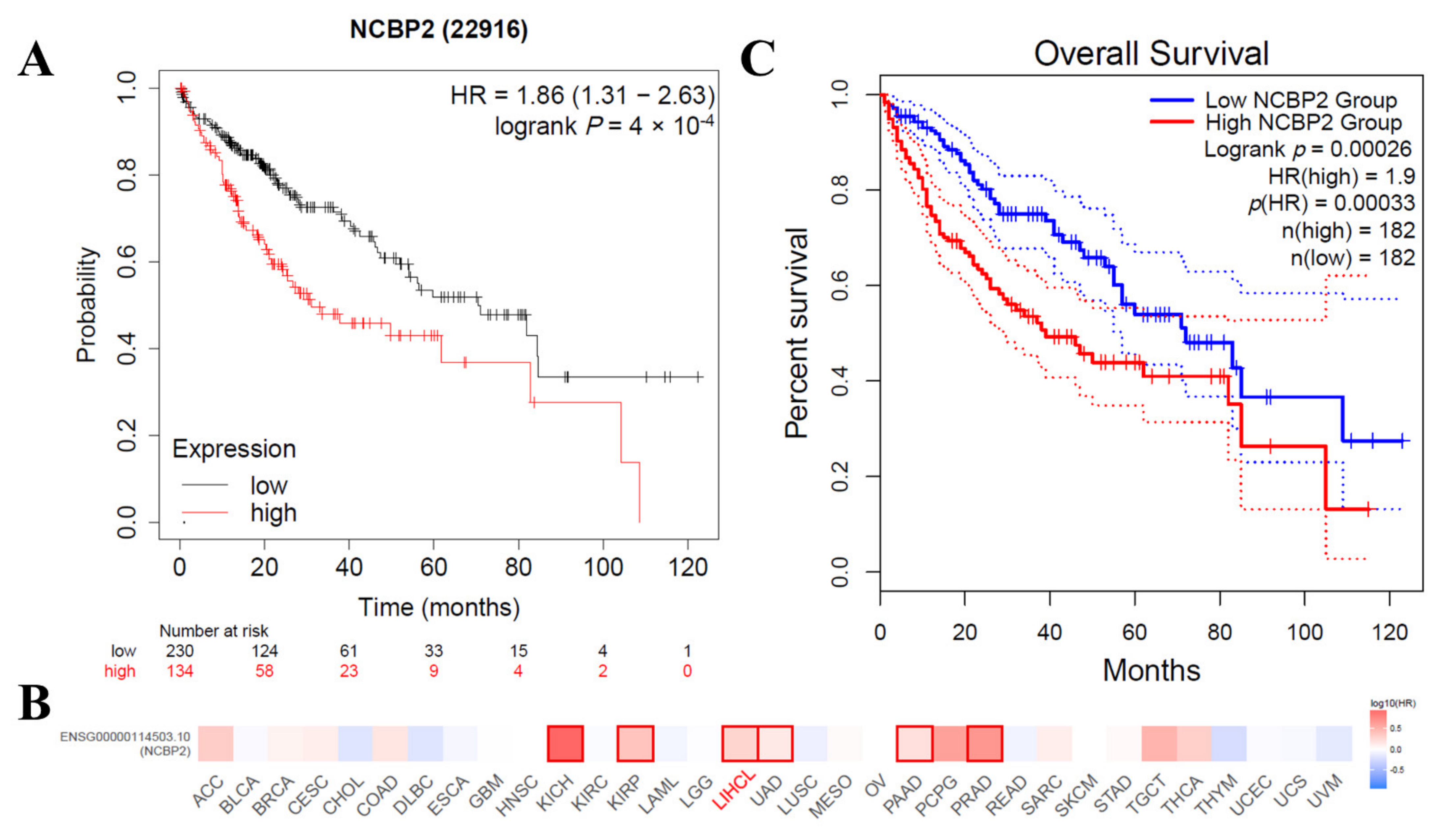
3.6. Correlations between the mRNA Expression Level of NCBP2 and Survival in HCC Patients
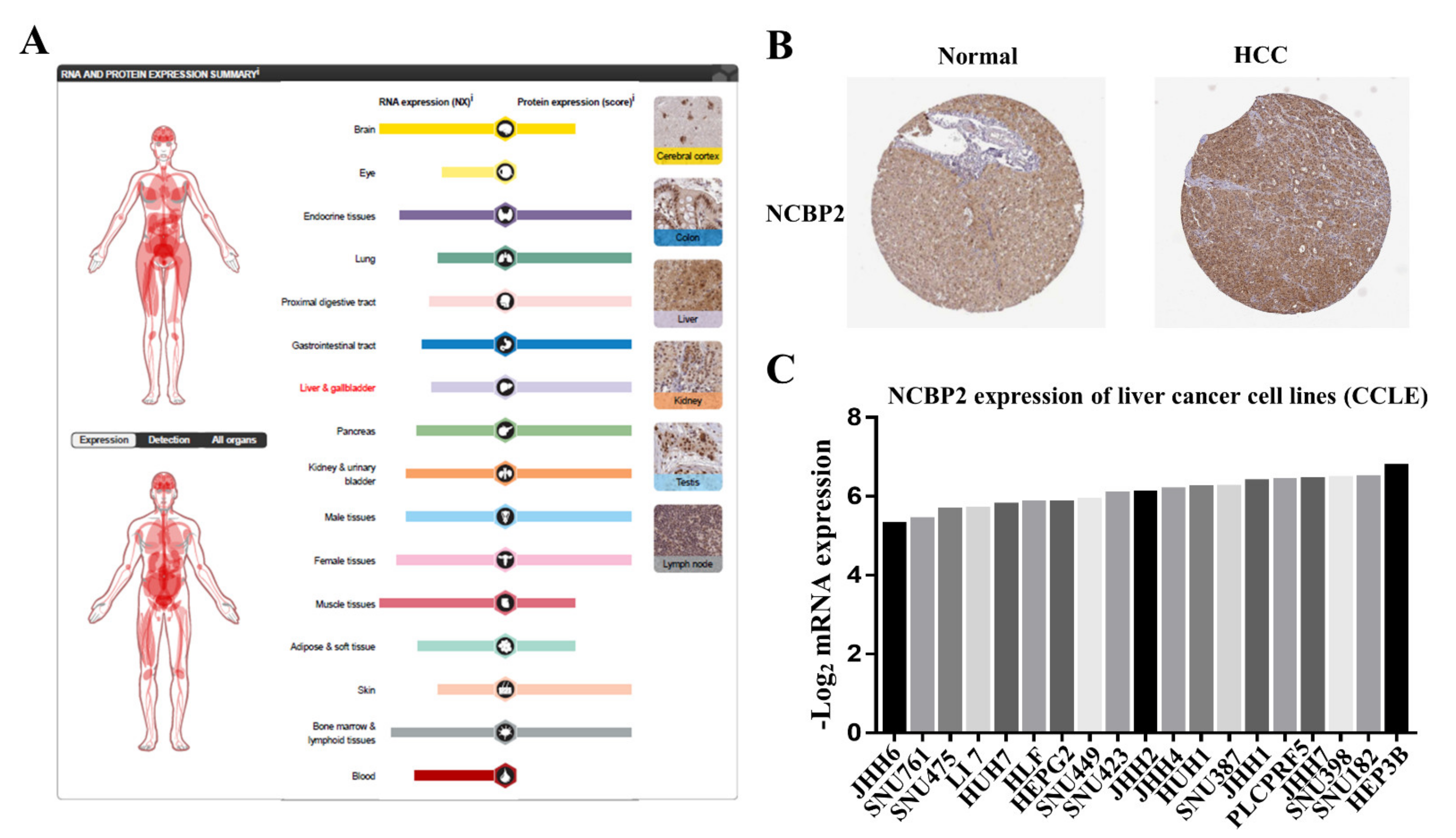
3.7. Protein Expression Level of NCBP2 in Human Tissue and Cell Lines
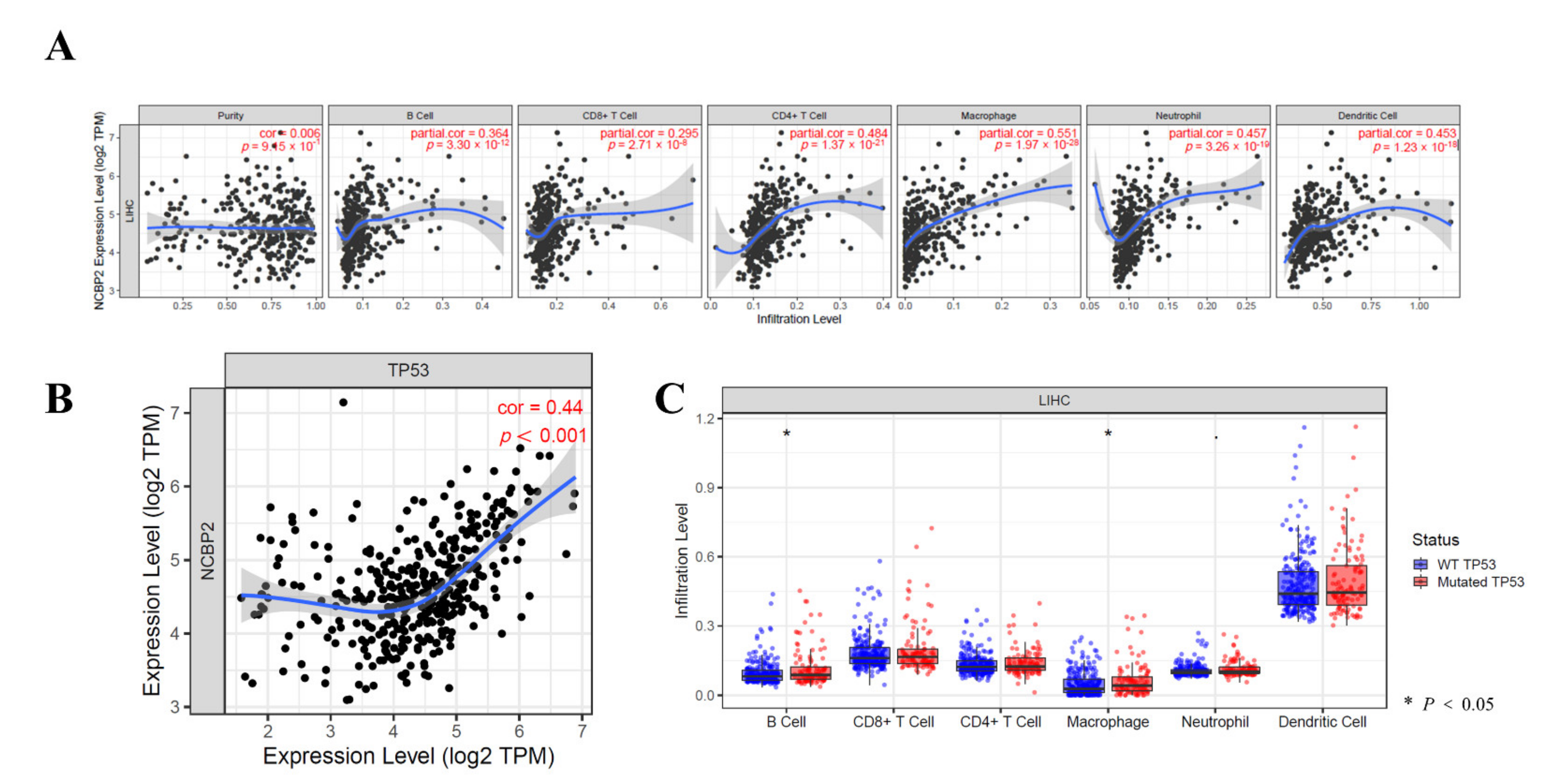
3.8. Relationship between the NCBP2 Expression and TP53 Mutation with Immune Makers
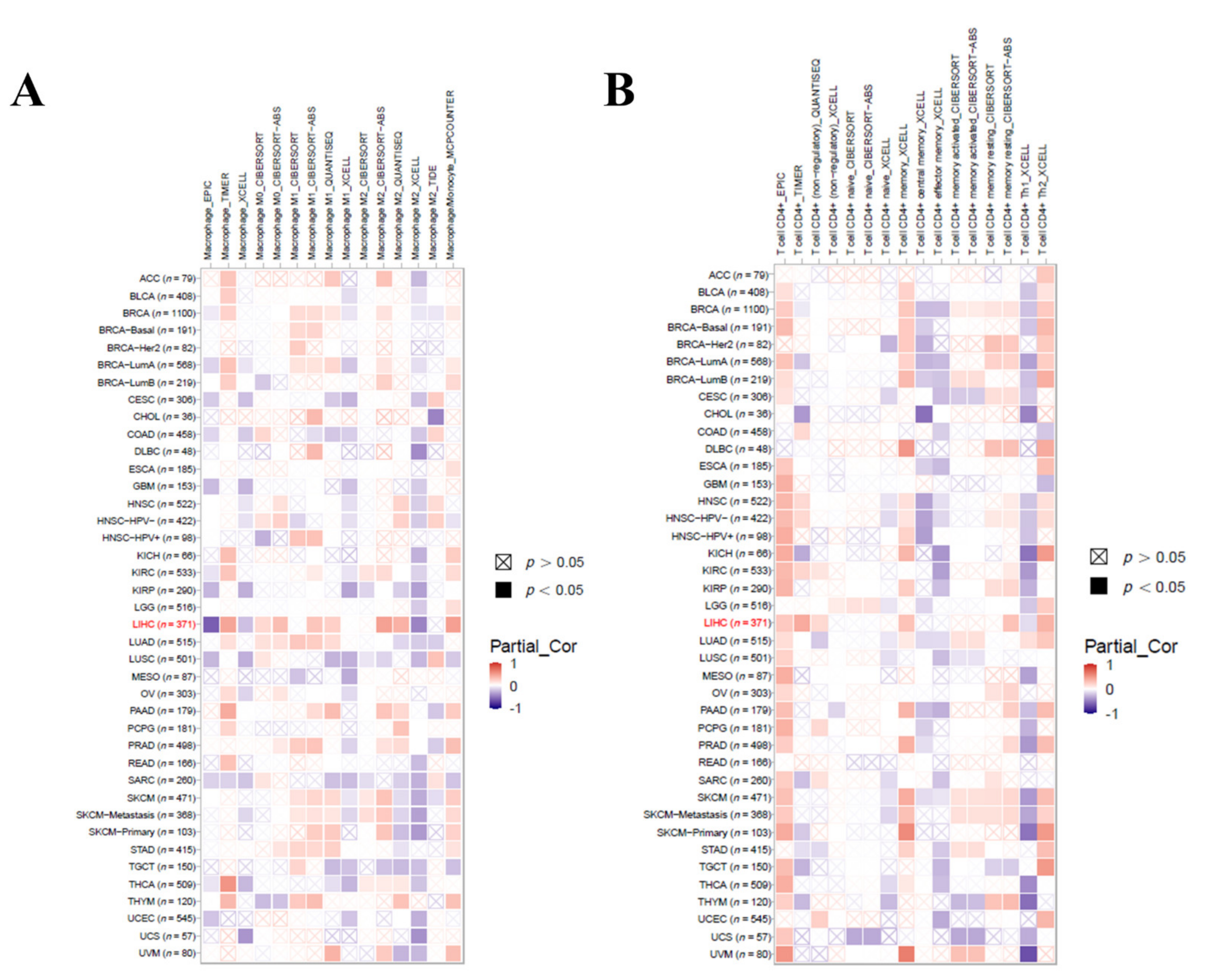
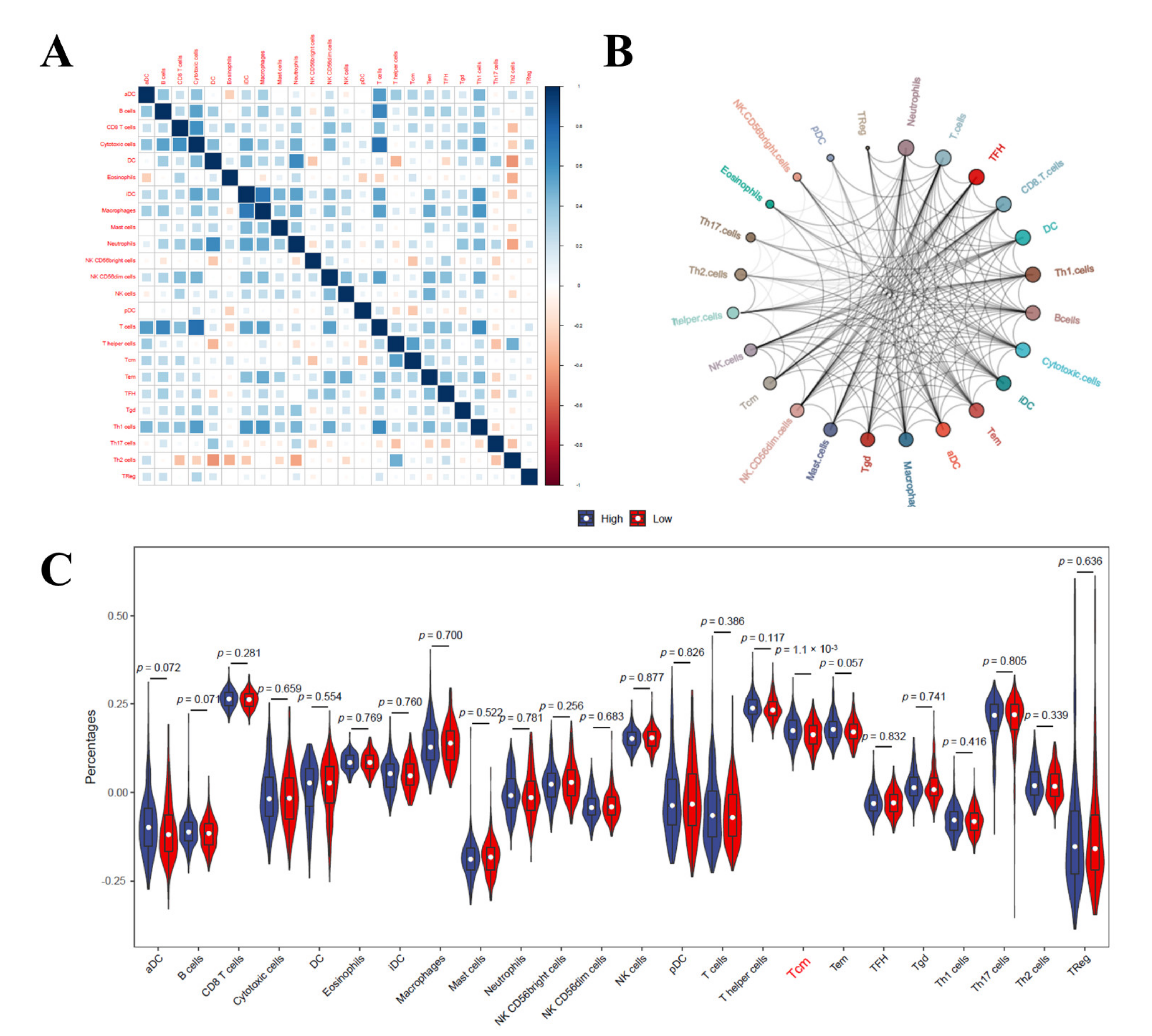
3.9. Immune Cell Infiltration Analysis in LIHC
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. 1), 4–13. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Singal, A.G. Surveillance for Hepatocellular Carcinoma: Current Best Practice and Future Direction. Gastroenterology 2019, 157, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Balague-Dobon, L.; Caceres, A.; Gonzalez, J.R. Fully exploiting SNP arrays: A systematic review on the tools to extract underlying genomic structure. Brief. Bioinform. 2022, 23, bbac043. [Google Scholar] [CrossRef]
- Vokes, N.I.; Chambers, E.; Nguyen, T.; Coolidge, A.; Lydon, C.A.; Le, X.; Sholl, L.; Heymach, J.V.; Nishino, M.; Van Allen, E.M.; et al. Concurrent TP53 mutations facilitate resistance evolution in EGFR mutant lung adenocarcinoma. J. Thorac. Oncol. 2022, 17, 779–792. [Google Scholar] [CrossRef]
- Jang, J.W.; Kim, J.S.; Kim, H.S.; Tak, K.Y.; Lee, S.K.; Nam, H.C.; Sung, P.S.; Kim, C.M.; Park, J.Y.; Bae, S.H.; et al. Significance of TERT Genetic Alterations and Telomere Length in Hepatocellular Carcinoma. Cancers 2021, 13, 2160. [Google Scholar] [CrossRef]
- Shao, W.; Ding, Q.; Guo, Y.; Xing, J.; Huo, Z.; Wang, Z.; Xu, Q.; Guo, Y. A Pan-Cancer Landscape of HOX-Related lncRNAs and Their Association with Prognosis and Tumor Microenvironment. Front. Mol. Biosci. 2021, 8, 767856. [Google Scholar] [CrossRef]
- McGrail, D.J.; Pilie, P.G.; Rashid, N.U.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.B.; Lim, B.; et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 2021, 32, 661–672. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Klempner, S.J.; Fabrizio, D.; Bane, S.; Reinhart, M.; Peoples, T.; Ali, S.M.; Sokol, E.S.; Frampton, G.; Schrock, A.B.; Anhorn, R.; et al. Tumor Mutational Burden as a Predictive Biomarker for Response to Immune Checkpoint Inhibitors: A Review of Current Evidence. Oncologist 2020, 25, e147–e159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, Z.J.; Heinrich, B.; Greten, T.F. Mouse models of hepatocellular carcinoma: An overview and highlights for immunotherapy research. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 536–554. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Z.; Liu, L.; Guo, C.; Jiao, D.; Li, L.; Zhao, J.; Han, X.; Sun, Y. CELF2 is a candidate prognostic and immunotherapy biomarker in triple-negative breast cancer and lung squamous cell carcinoma: A pan-cancer analysis. J. Cell Mol. Med. 2021, 25, 7559–7574. [Google Scholar] [CrossRef]
- Teo, M.Y.; Seier, K.; Ostrovnaya, I.; Regazzi, A.M.; Kania, B.E.; Moran, M.M.; Cipolla, C.K.; Bluth, M.J.; Chaim, J.; Al-Ahmadie, H.; et al. Alterations in DNA Damage Response and Repair Genes as Potential Marker of Clinical Benefit From PD-1/PD-L1 Blockade in Advanced Urothelial Cancers. J. Clin. Oncol. 2018, 36, 1685–1694. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Kim, H.N.; Newcomb, C.W.; Carbonari, D.M.; Roy, J.A.; Torgersen, J.; Althoff, K.N.; Kitahata, M.M.; Reddy, K.R.; Lim, J.K.; Silverberg, M.J.; et al. Risk of HCC with Hepatitis B Viremia Among HIV/HBV-Coinfected Persons in North America. Hepatology 2021, 74, 1190–1202. [Google Scholar] [CrossRef]
- Kanwal, F.; Khaderi, S.; Singal, A.G.; Marrero, J.A.; Loo, N.; Asrani, S.K.; Amos, C.I.; Thrift, A.P.; Gu, X.; Luster, M.; et al. Risk Factors for Hepatocellular Cancer in Contemporary Cohorts of Patients with Cirrhosis. Hepatology 2022. [Google Scholar] [CrossRef]
- Ahn, J.C.; Teng, P.C.; Chen, P.J.; Posadas, E.; Tseng, H.R.; Lu, S.C.; Yang, J.D. Detection of Circulating Tumor Cells and Their Implications as a Biomarker for Diagnosis, Prognostication, and Therapeutic Monitoring in Hepatocellular Carcinoma. Hepatology 2021, 73, 422–436. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D. PD-L1, TMB, and other potential predictors of response to immunotherapy for hepatocellular carcinoma: How can they assist drug clinical trials? Expert Opin. Investig. Drugs 2021, 31, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Tan, J.; Wu, M.; Fan, W.; Wei, J.; Zhu, B.; Guo, J.; Wang, S.; Zhou, P.; Zhang, H.; et al. High-affinity neoantigens correlate with better prognosis and trigger potent antihepatocellular carcinoma (HCC) activity by activating CD39(+)CD8(+) T cells. Gut 2021, 70, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Bersanelli, M. Tumour mutational burden as a driver for treatment choice in resistant tumours (and beyond). Lancet Oncol. 2020, 21, 1255–1257. [Google Scholar] [CrossRef]
- Nataraj, N.B.; Noronha, A.; Lee, J.S.; Ghosh, S.; Mohan, R.H.; Sekar, A.; Zuckerman, B.; Lindzen, M.; Tarcitano, E.; Srivastava, S.; et al. Nucleoporin-93 reveals a common feature of aggressive breast cancers: Robust nucleocytoplasmic transport of transcription factors. Cell Rep. 2022, 38, 110418. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, Y.; Ren, S.; Xiao, X.; Cao, H.; He, J. A Pan-Cancer Analysis of the Oncogenic Role of Nuclear Transport Factor 2 in Human Cancers. Front. Oncol. 2022, 12, 829389. [Google Scholar] [CrossRef]
- Zhong, F.J.; Sun, B.; Cao, M.M.; Xu, C.; Li, Y.M.; Yang, L.Y. STMN2 mediates nuclear translocation of Smad2/3 and enhances TGFbeta signaling by destabilizing microtubules to promote epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2021, 506, 128–141. [Google Scholar] [CrossRef]
- Feng, J.; Guo, J.; Zhao, P.; Shen, J.; Chai, B.; Wang, J. mTOR up-regulation of SNRPA1 contributes to hepatocellular carcinoma development. Biosci. Rep. 2020, 40, BSR20193815. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, Z.; Chen, L.; Li, Q.; Zou, J.; Liu, S. PPM1G promotes the progression of hepatocellular carcinoma via phosphorylation regulation of alternative splicing protein SRSF3. Cell Death Dis. 2021, 12, 722. [Google Scholar] [CrossRef]
- Xue, C.; Zhao, Y.; Li, G.; Li, L. Multi-Omic Analyses of the m(5)C Regulator ALYREF Reveal Its Essential Roles in Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 633415. [Google Scholar] [CrossRef]
- Behary, J.; Amorim, N.; Jiang, X.T.; Raposo, A.; Gong, L.; McGovern, E.; Ibrahim, R.; Chu, F.; Stephens, C.; Jebeili, H.; et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat. Commun. 2021, 12, 187. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, X.; Peng, Z.; Li, C.; Wang, Z.; Wang, C.; Deng, Z.; Wu, B.; Cui, Y.; Wang, Z.; et al. Deubiquitylase OTUD6B Governs pVHL Stability in an Enzyme-Independent Manner and Suppresses Hepatocellular Carcinoma Metastasis. Adv. Sci. 2020, 7, 1902040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.D.; Fang, L.; Yu, H.Q.; Zhang, J.; Lin, X.T.; Liu, X.Y.; Wu, D.; Li, G.X.; Huang, D.; Zhang, Y.J.; et al. p53 haploinsufficiency and increased mTOR signalling define a subset of aggressive hepatocellular carcinoma. J. Hepatol. 2021, 74, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.D.; Jensen, M.; Lasser, M.; Huber, E.; Yusuff, T.; Pizzo, L.; Lifschutz, B.; Desai, I.; Kubina, A.; Yennawar, S.; et al. NCBP2 modulates neurodevelopmental defects of the 3q29 deletion in Drosophila and Xenopus laevis models. PLoS Genet. 2020, 16, e1008590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebhardt, A.; Bergant, V.; Schnepf, D.; Moser, M.; Meiler, A.; Togbe, D.; Mackowiak, C.; Reinert, L.S.; Paludan, S.R.; Ryffel, B.; et al. The alternative cap-binding complex is required for antiviral defense in vivo. PLoS Pathog. 2019, 15, e1008155. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Jiang, Q.; Liu, L.; Peng, H.; Wang, Y.; Li, S.; Tang, Y.; Yu, J.; Yang, J.; Liu, Z. Integrated Analysis of RNA-Binding Proteins Associated With the Prognosis and Immunosuppression in Squamous Cell Carcinoma of Head and Neck. Front. Genet. 2020, 11, 571403. [Google Scholar] [CrossRef]
- Nastase, A.; Lupo, A.; Laszlo, V.; Damotte, D.; Dima, S.; Canny, E.; Alifano, M.; Popescu, I.; Klepetko, W.; Grigoroiu, M. Platinum Drug Sensitivity Polymorphisms in Stage III Non-small Cell Lung Cancer with Invasion of Mediastinal Lymph Nodes. Cancer Genom. Proteom. 2020, 17, 587–595. [Google Scholar] [CrossRef]
- Wang, L.L.; Yan, D.; Tang, X.; Zhang, M.; Liu, S.; Wang, Y.; Zhang, M.; Zhou, G.; Li, T.; Jiang, F.; et al. High Expression of BCL11A Predicts Poor Prognosis for Childhood MLL-r ALL. Front. Oncol. 2021, 11, 755188. [Google Scholar] [CrossRef]
- Zhang, H.; Cheng, L.; Liu, C. Regulatory Networks of Prognostic mRNAs in Pediatric Acute Myeloid Leukemia. J. Healthc. Eng. 2022, 2022, 2691997. [Google Scholar] [CrossRef]
- Kugeratski, F.G.; Atkinson, S.J.; Neilson, L.J.; Lilla, S.; Knight, J.; Serneels, J.; Juin, A.; Ismail, S.; Bryant, D.M.; Markert, E.K.; et al. Hypoxic cancer-associated fibroblasts increase NCBP2-AS2/HIAR to promote endothelial sprouting through enhanced VEGF signaling. Sci. Signal. 2019, 12, eaan8247. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.Y.; Yang, W.; Zheng, F.S.; Wang, Y.B.; Lu, J.B. Long non-coding RNA SNHG1 regulates zinc finger E-box binding homeobox 1 expression by interacting with TAp63 and promotes cell metastasis and invasion in Lung squamous cell carcinoma. Biomed. Pharmacother. 2017, 90, 650–658. [Google Scholar] [CrossRef]
- Xu, W.; Deng, B.; Lin, P.; Liu, C.; Li, B.; Huang, Q.; Zhou, H.; Yang, J.; Qu, L. Ribosome profiling analysis identified a KRAS-interacting microprotein that represses oncogenic signaling in hepatocellular carcinoma cells. Sci. China Life Sci. 2020, 63, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Lin, Z.; Liu, L.; Zhang, R.; Geng, Y.; Fan, M.; Zhu, W.; Lu, M.; Lu, L.; Jia, H.; et al. GOLM1 exacerbates CD8(+) T cell suppression in hepatocellular carcinoma by promoting exosomal PD-L1 transport into tumor-associated macrophages. Signal Transduct. Target. Ther. 2021, 6, 397. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Mei, J.; Yi, S.; Feng, C.; Ma, Y.; Liu, Y.; Liu, Y.; Chen, C. Tumor associated macrophage and microbe: The potential targets of tumor vaccine delivery. Adv. Drug Deliv. Rev. 2022, 180, 114046. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Zhu, J.; Zhao, Z.; Lu, C.; Liu, S.; Fang, S.; Zheng, L.; Zhang, N.; Chen, M.; Xu, M.; et al. Diagnosis and prognosis models for hepatocellular carcinoma patient’s management based on tumor mutation burden. J. Adv. Res. 2021, 33, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Wang, D.; Qiu, X.; Luo, G.; Wu, T.; Yang, S.; Li, Z.; Zhu, Y.; Wang, S.; Wu, R.; et al. Trajectory and Functional Analysis of PD-1(high) CD4(+)CD8(+) T Cells in Hepatocellular Carcinoma by Single-Cell Cytometry and Transcriptome Sequencing. Adv. Sci. 2020, 7, 2000224. [Google Scholar] [CrossRef]
- Park, S.L.; Gebhardt, T.; Mackay, L.K. Tissue-Resident Memory T Cells in Cancer Immunosurveillance. Trends Immunol. 2019, 40, 735–747. [Google Scholar] [CrossRef]
- Okla, K.; Farber, D.L.; Zou, W. Tissue-resident memory T cells in tumor immunity and immunotherapy. J. Exp. Med. 2021, 218, e20201605. [Google Scholar] [CrossRef]
- Pinato, D.J.; Guerra, N.; Fessas, P.; Murphy, R.; Mineo, T.; Mauri, F.A.; Mukherjee, S.K.; Thursz, M.; Wong, C.N.; Sharma, R.; et al. Immune-based therapies for hepatocellular carcinoma. Oncogene 2020, 39, 3620–3637. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Univariate Cox | Multivariate Cox | ||
|---|---|---|---|---|
| HR (95% CI) | p Value | HR (95% CI) | p Value | |
| stage2 | 1.576 | 0.345 | 1.367 | 0.215 |
| stage3 | 2.205 | 0.001 ** | 2.205 | 0.001 ** |
| stage4 | 4.575 | 0.005 * | 4.575 | 0.015 * |
| Gender male | 0.907 | 0.789 | 0.907 | 0.632 |
| B_cell | 0.008 | 0.235 | 0.008 | 0.182 |
| CD8+ _ T cell | 0.005 | 0.045 * | 0.005 | 0.037 * |
| CD4+ _ T cell | 0.014 | 0.34 | 0.014 | 0.22 |
| NCBP2 | 1.467 | 0.006 ** | 1.437 | 0.046 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Huang, T.; Huang, P. Integrated Analysis of Tumor Mutation Burden and Immune Infiltrates in Hepatocellular Carcinoma. Diagnostics 2022, 12, 1918. https://doi.org/10.3390/diagnostics12081918
Zhao Y, Huang T, Huang P. Integrated Analysis of Tumor Mutation Burden and Immune Infiltrates in Hepatocellular Carcinoma. Diagnostics. 2022; 12(8):1918. https://doi.org/10.3390/diagnostics12081918
Chicago/Turabian StyleZhao, Yulan, Ting Huang, and Pintong Huang. 2022. "Integrated Analysis of Tumor Mutation Burden and Immune Infiltrates in Hepatocellular Carcinoma" Diagnostics 12, no. 8: 1918. https://doi.org/10.3390/diagnostics12081918
APA StyleZhao, Y., Huang, T., & Huang, P. (2022). Integrated Analysis of Tumor Mutation Burden and Immune Infiltrates in Hepatocellular Carcinoma. Diagnostics, 12(8), 1918. https://doi.org/10.3390/diagnostics12081918