
Pathological and Molecular Features of Nodal Peripheral T-Cell Lymphomas


Abstract
:1. Introduction
2. ALCL
2.1. ALK+ ALCL
2.1.1. Clinical Features
2.1.2. Morphological and Immunohistochemical Features
2.1.3. Molecular Features
2.2. ALK− ALCL
2.2.1. DUSP22-Rearranged, TP63-Rearranged, and Triple-Negative ALCL
2.2.2. Clinical Features
2.2.3. Morphological and Immunohistochemical Features
2.2.4. Molecular Features
3. Nodal T-Cell Lymphoma with TFH Cell Origin
3.1. AITL
3.1.1. Clinical Features
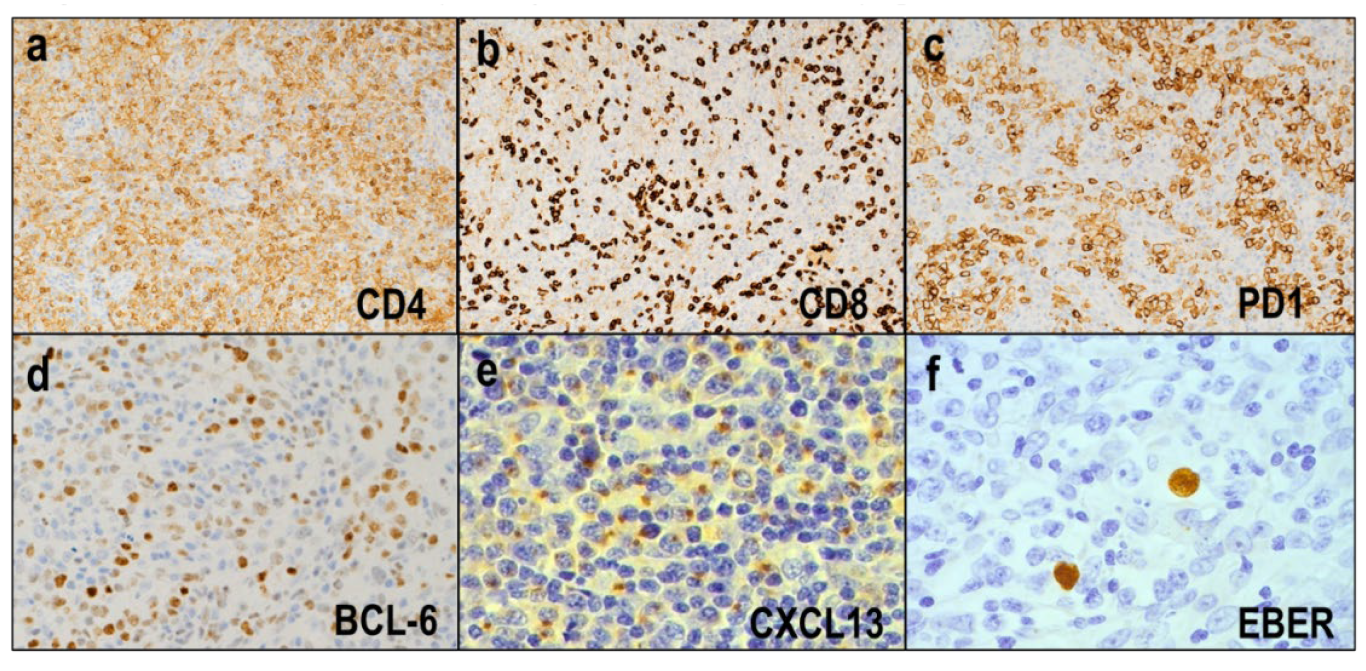
3.1.2. Morphological and Immunohistochemical Features
3.1.3. Molecular Features
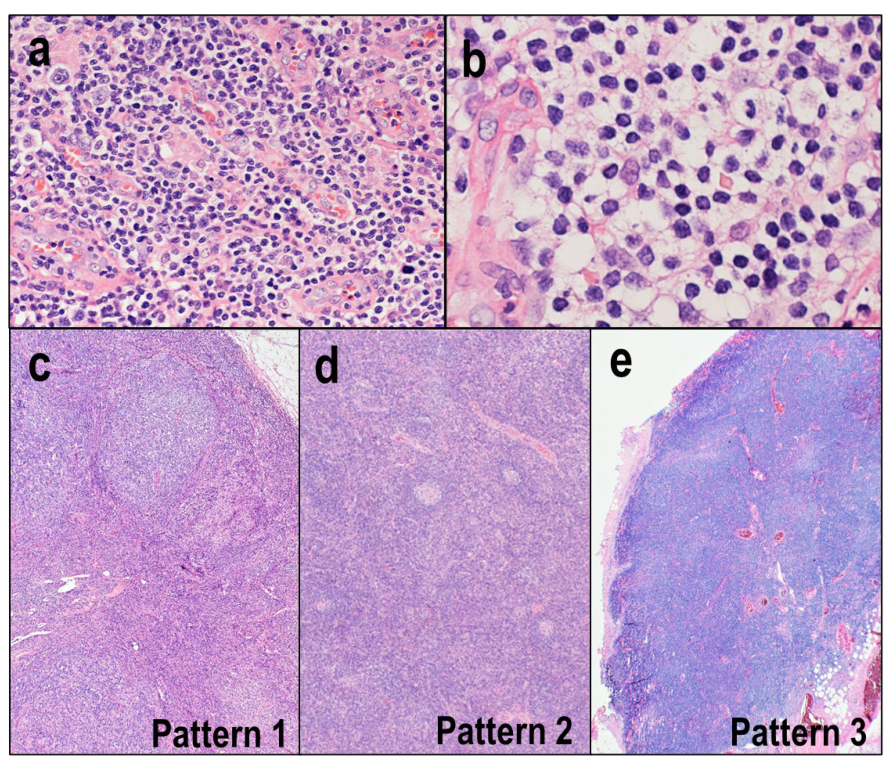
3.2. FTCL and nPTCL-TFH
3.2.1. Clinical Features
3.2.2. Morphological and Immunohistochemical Features
3.2.3. Molecular Features
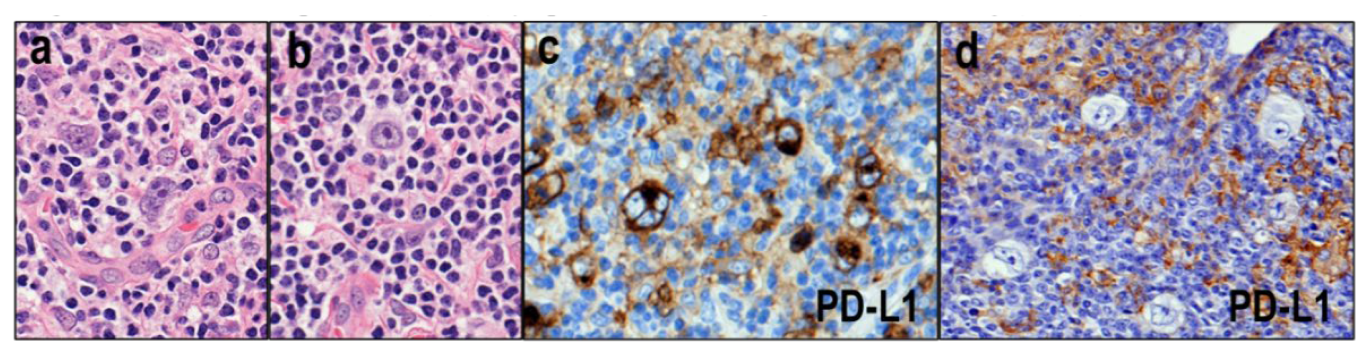
3.3. TFH Lymphomas with Hodgkin–Reed–Sternberg (HRS)-Like Cells
4. ATLL
4.1. Clinical Features
4.2. Morphological and Immunohistochemical Features
4.3. Molecular and Microenvironmental Features
5. PTCL-NOS
5.1. Clinical Features
5.2. Morphological and Immunohistochemical Features
5.3. Molecular and Microenvironmental Features
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Swerdlow, S.; Campo, E.; Harris, N.; Jaffe, E.; Pileri, S.; Stein, H.; Thiele, J. (Eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017. [Google Scholar]
- Vega, F.; Amador, C.; Chadburn, A.; Hsi, E.D.; Slack, G.; Medeiros, L.J.; Feldman, A.L. Genetic profiling and biomarkers in peripheral T-cell lymphomas: Current role in the diagnostic work-up. Mod. Pathol. Off. J. USA Can. Acad. Pathol. Inc. 2022, 35, 306–318. [Google Scholar] [CrossRef]
- Chihara, D.; Ito, H.; Matsuda, T.; Shibata, A.; Katsumi, A.; Nakamura, S.; Tomotaka, S.; Morton, L.M.; Weisenburger, D.D.; Matsuo, K. Differences in incidence and trends of haematological malignancies in Japan and the United States. Br. J. Haematol. 2014, 164, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Pileri, S.A.; Tabanelli, V.; Fiori, S.; Calleri, A.; Melle, F.; Motta, G.; Lorenzini, D.; Tarella, C.; Derenzini, E. Peripheral T-Cell Lymphoma, Not Otherwise Specified: Clinical Manifestations, Diagnosis, and Future Treatment. Cancers 2021, 13, 4535. [Google Scholar] [CrossRef]
- Willemze, R.; Jaffe, E.S.; Burg, G.; Cerroni, L.; Berti, E.; Swerdlow, S.H.; Ralfkiaer, E.; Chimenti, S.; Diaz-Perez, J.L.; Duncan, L.M.; et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005, 105, 3768–3785. [Google Scholar] [CrossRef] [PubMed]
- Bekkenk, M.W.; Geelen, F.A.; van Voorst Vader, P.C.; Heule, F.; Geerts, M.L.; van Vloten, W.A.; Meijer, C.J.; Willemze, R. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: A report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood 2000, 95, 3653–3661. [Google Scholar] [CrossRef]
- Oishi, N.; Miranda, R.N.; Feldman, A.L. Genetics of Breast Implant-Associated Anaplastic Large Cell Lymphoma (BIA-ALCL). Aesthetic Surg. J. 2019, 39, S14–S20. [Google Scholar] [CrossRef]
- Amador, C.; Feldman, A.L. How I Diagnose Anaplastic Large Cell Lymphoma. Am. J. Clin. Pathol. 2021, 155, 479–497. [Google Scholar] [CrossRef]
- Hapgood, G.; Savage, K.J. The biology and management of systemic anaplastic large cell lymphoma. Blood 2015, 126, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Benharroch, D.; Meguerian-Bedoyan, Z.; Lamant, L.; Amin, C.; Brugieres, L.; Terrier-Lacombe, M.J.; Haralambieva, E.; Pulford, K.; Pileri, S.; Morris, S.W.; et al. ALK-positive lymphoma: A single disease with a broad spectrum of morphology. Blood 1998, 91, 2076–2084. [Google Scholar] [CrossRef] [PubMed]
- Lamant, L.; McCarthy, K.; d’Amore, E.; Klapper, W.; Nakagawa, A.; Fraga, M.; Maldyk, J.; Simonitsch-Klupp, I.; Oschlies, I.; Delsol, G.; et al. Prognostic impact of morphologic and phenotypic features of childhood ALK-positive anaplastic large-cell lymphoma: Results of the ALCL99 study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 4669–4676. [Google Scholar] [CrossRef]
- Spiegel, A.; Paillard, C.; Ducassou, S.; Perel, Y.; Plantaz, D.; Strullu, M.; Eischen, A.; Lutz, P.; Lamant, L.; Le Deley, M.C.; et al. Paediatric anaplastic large cell lymphoma with leukaemic presentation in children: A report of nine French cases. Br. J. Haematol. 2014, 165, 545–551. [Google Scholar] [CrossRef]
- Kinney, M.C.; Collins, R.D.; Greer, J.P.; Whitlock, J.A.; Sioutos, N.; Kadin, M.E. A small-cell-predominant variant of primary Ki-1 (CD30)+ T-cell lymphoma. Am. J. Surg. Pathol. 1993, 17, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Satou, A.; Asano, N.; Tatekawa, S.; Fukuyama, R.; Nakamura, S. Lymphohistiocytic and small cell pattern of anaplastic large cell lymphoma, ALK positive, arising in an 86-year-old woman. Pathol. Int. 2013, 63, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, J.; Lamant, L.; Brugieres, L.; Gaillard, F.; Campo, E.; Brousset, P.; Delsol, G. ALK-positive anaplastic large cell lymphoma mimicking nodular sclerosis Hodgkin’s lymphoma: Report of 10 cases. Am. J. Surg. Pathol. 2006, 30, 223–229. [Google Scholar] [CrossRef]
- Chan, J.K.; Buchanan, R.; Fletcher, C.D. Sarcomatoid variant of anaplastic large-cell Ki-1 lymphoma. Am. J. Surg. Pathol. 1990, 14, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.S. Anaplastic large cell lymphoma: The shifting sands of diagnostic hematopathology. Mod. Pathol. Off. J. USA Can. Acad. Pathol. Inc. 2001, 14, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Foss, H.D.; Anagnostopoulos, I.; Araujo, I.; Assaf, C.; Demel, G.; Kummer, J.A.; Hummel, M.; Stein, H. Anaplastic large-cell lymphomas of T-cell and null-cell phenotype express cytotoxic molecules. Blood 1996, 88, 4005–4011. [Google Scholar] [CrossRef] [PubMed]
- de Leval, L. Approach to nodal-based T-cell lymphomas. Pathology 2020, 52, 78–99. [Google Scholar] [CrossRef] [PubMed]
- Boi, M.; Zucca, E.; Inghirami, G.; Bertoni, F. Advances in understanding the pathogenesis of systemic anaplastic large cell lymphomas. Br. J. Haematol. 2015, 168, 771–783. [Google Scholar] [CrossRef]
- Feldman, A.L.; Dogan, A.; Smith, D.I.; Law, M.E.; Ansell, S.M.; Johnson, S.H.; Porcher, J.C.; Ozsan, N.; Wieben, E.D.; Eckloff, B.W.; et al. Discovery of recurrent t(6; 7)(p25.3; q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood 2011, 117, 915–919. [Google Scholar] [CrossRef]
- Parrilla Castellar, E.R.; Jaffe, E.S.; Said, J.W.; Swerdlow, S.H.; Ketterling, R.P.; Knudson, R.A.; Sidhu, J.S.; Hsi, E.D.; Karikehalli, S.; Jiang, L.; et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood 2014, 124, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Karube, K.; Feldman, A.L. “Double-hit” of DUSP22 and TP63 rearrangements in anaplastic large cell lymphoma, ALK-negative. Blood 2020, 135, 700. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.J.; Harris, N.L.; Vose, J.M.; Ullrich, F.; Jaffe, E.S.; Connors, J.M.; Rimsza, L.; Pileri, S.A.; Chhanabhai, M.; Gascoyne, R.D.; et al. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: Report from the International Peripheral T-Cell Lymphoma Project. Blood 2008, 111, 5496–5504. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.B.; Hamilton-Dutoit, S.J.; Bendix, K.; Ketterling, R.P.; Bedroske, P.P.; Luoma, I.M.; Sattler, C.A.; Boddicker, R.L.; Bennani, N.N.; Norgaard, P.; et al. DUSP22 and TP63 rearrangements predict outcome of ALK-negative anaplastic large cell lymphoma: A Danish cohort study. Blood 2017, 130, 554–557. [Google Scholar] [CrossRef] [PubMed]
- King, R.L.; Dao, L.N.; McPhail, E.D.; Jaffe, E.S.; Said, J.; Swerdlow, S.H.; Sattler, C.A.; Ketterling, R.P.; Sidhu, J.S.; Hsi, E.D.; et al. Morphologic Features of ALK-negative Anaplastic Large Cell Lymphomas With DUSP22 Rearrangements. Am. J. Surg. Pathol. 2016, 40, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, A.; Feldman, A.L.; Ketterling, R.P.; Dasari, S.; Rech, K.L.; McPhail, E.D.; Kurtin, P.J.; Shi, M. Striking Association of Lymphoid Enhancing Factor (LEF1) Overexpression and DUSP22 Rearrangements in Anaplastic Large Cell Lymphoma. Am. J. Surg. Pathol. 2021, 45, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Boddicker, R.L.; Dasari, S.; Sidhu, J.S.; Kadin, M.E.; Macon, W.R.; Ansell, S.M.; Ketterling, R.P.; Rech, K.L.; Feldman, A.L. Expression of p63 protein in anaplastic large cell lymphoma: Implications for genetic subtyping. Hum. Pathol. 2017, 64, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Luchtel, R.A.; Dasari, S.; Oishi, N.; Pedersen, M.B.; Hu, G.; Rech, K.L.; Ketterling, R.P.; Sidhu, J.; Wang, X.; Katoh, R.; et al. Molecular profiling reveals immunogenic cues in anaplastic large cell lymphomas with DUSP22 rearrangements. Blood 2018, 132, 1386–1398. [Google Scholar] [CrossRef]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef] [PubMed]
- Luchtel, R.A.; Zimmermann, M.T.; Hu, G.; Dasari, S.; Jiang, M.; Oishi, N.; Jacobs, H.K.; Zeng, Y.; Hundal, T.; Rech, K.L.; et al. Recurrent MSC (E116K) mutations in ALK-negative anaplastic large cell lymphoma. Blood 2019, 133, 2776–2789. [Google Scholar] [CrossRef]
- de Leval, L.; Parrens, M.; Le Bras, F.; Jais, J.P.; Fataccioli, V.; Martin, A.; Lamant, L.; Delarue, R.; Berger, F.; Arbion, F.; et al. Angioimmunoblastic T-cell lymphoma is the most common T-cell lymphoma in two distinct French information data sets. Haematologica 2015, 100, e361–e364. [Google Scholar] [CrossRef] [PubMed]
- Vose, J.; Armitage, J.; Weisenburger, D.; International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: Pathology findings and clinical outcomes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 4124–4130. [Google Scholar] [CrossRef]
- Federico, M.; Rudiger, T.; Bellei, M.; Nathwani, B.N.; Luminari, S.; Coiffier, B.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Savage, K.J.; et al. Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: Analysis of the international peripheral T-cell lymphoma project. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, T.; Shimada, K.; Yamamoto, K.; Chihara, D.; Ichihashi, T.; Oshima, R.; Tanimoto, M.; Iwasaki, T.; Isoda, A.; Sakai, A.; et al. Retrospective analysis of prognostic factors for angioimmunoblastic T-cell lymphoma: A multicenter cooperative study in Japan. Blood 2012, 119, 2837–2843. [Google Scholar] [CrossRef] [PubMed]
- Mourad, N.; Mounier, N.; Briere, J.; Raffoux, E.; Delmer, A.; Feller, A.; Meijer, C.J.; Emile, J.F.; Bouabdallah, R.; Bosly, A.; et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte (GELA) trials. Blood 2008, 111, 4463–4470. [Google Scholar] [CrossRef] [PubMed]
- Attygalle, A.D.; Kyriakou, C.; Dupuis, J.; Grogg, K.L.; Diss, T.C.; Wotherspoon, A.C.; Chuang, S.S.; Cabecadas, J.; Isaacson, P.G.; Du, M.Q.; et al. Histologic evolution of angioimmunoblastic T-cell lymphoma in consecutive biopsies: Clinical correlation and insights into natural history and disease progression. Am. J. Surg. Pathol. 2007, 31, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Gaulard, P.; de Leval, L. Follicular helper T cells: Implications in neoplastic hematopathology. Semin. Diagn. Pathol. 2011, 28, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Basha, B.M.; Bryant, S.C.; Rech, K.L.; Feldman, A.L.; Vrana, J.A.; Shi, M.; Reed, K.A.; King, R.L. Application of a 5 Marker Panel to the Routine Diagnosis of Peripheral T-Cell Lymphoma with T-Follicular Helper Phenotype. Am. J. Surg. Pathol. 2019, 43, 1282–1290. [Google Scholar] [CrossRef]
- Eladl, A.E.; Shimada, K.; Suzuki, Y.; Takahara, T.; Kato, S.; Kohno, K.; Elsayed, A.A.; Wu, C.C.; Tokunaga, T.; Kinoshita, T.; et al. EBV status has prognostic implication among young patients with angioimmunoblastic T-cell lymphoma. Cancer Med. 2020, 9, 678–688. [Google Scholar] [CrossRef]
- Zettl, A.; Lee, S.S.; Rudiger, T.; Starostik, P.; Marino, M.; Kirchner, T.; Ott, M.; Muller-Hermelink, H.K.; Ott, G. Epstein-Barr virus-associated B-cell lymphoproliferative disorders in angloimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma, unspecified. Am. J. Clin. Pathol. 2002, 117, 368–379. [Google Scholar] [CrossRef]
- Weiss, L.M.; Jaffe, E.S.; Liu, X.F.; Chen, Y.Y.; Shibata, D.; Medeiros, L.J. Detection and localization of Epstein-Barr viral genomes in angioimmunoblastic lymphadenopathy and angioimmunoblastic lymphadenopathy-like lymphoma. Blood 1992, 79, 1789–1795. [Google Scholar] [CrossRef]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef]
- Palomero, T.; Couronne, L.; Khiabanian, H.; Kim, M.Y.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Carpenter, Z.; Abate, F.; Allegretta, M.; Haydu, J.E.; et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 2014, 46, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, F.; Couronne, L.; Parrens, M.; Jais, J.P.; Travert, M.; Lamant, L.; Tournillac, O.; Rousset, T.; Fabiani, B.; Cairns, R.A.; et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood 2012, 120, 1466–1469. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Iqbal, J.; Lemonnier, F.; Kucuk, C.; de Leval, L.; Jais, J.P.; Parrens, M.; Martin, A.; Xerri, L.; Brousset, P.; et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 2012, 119, 1901–1903. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, F.; Cairns, R.A.; Inoue, S.; Li, W.Y.; Dupuy, A.; Broutin, S.; Martin, N.; Fataccioli, V.; Pelletier, R.; Wakeham, A.; et al. The IDH2 R172K mutation associated with angioimmunoblastic T-cell lymphoma produces 2HG in T cells and impacts lymphoid development. Proc. Natl. Acad. Sci. USA 2016, 113, 15084–15089. [Google Scholar] [CrossRef] [PubMed]
- Rohr, J.; Guo, S.; Huo, J.; Bouska, A.; Lachel, C.; Li, Y.; Simone, P.D.; Zhang, W.; Gong, Q.; Wang, C.; et al. Recurrent activating mutations of CD28 in peripheral T-cell lymphomas. Leukemia 2016, 30, 1062–1070. [Google Scholar] [CrossRef]
- Vallois, D.; Dobay, M.P.; Morin, R.D.; Lemonnier, F.; Missiaglia, E.; Juilland, M.; Iwaszkiewicz, J.; Fataccioli, V.; Bisig, B.; Roberti, A.; et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood 2016, 128, 1490–1502. [Google Scholar] [CrossRef]
- Fujisawa, M.; Sakata-Yanagimoto, M.; Nishizawa, S.; Komori, D.; Gershon, P.; Kiryu, M.; Tanzima, S.; Fukumoto, K.; Enami, T.; Muratani, M.; et al. Activation of RHOA-VAV1 signaling in angioimmunoblastic T-cell lymphoma. Leukemia 2018, 32, 694–702. [Google Scholar] [CrossRef]
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Sakata-Yanagimoto, M. Multistep tumorigenesis in peripheral T cell lymphoma. Int. J. Hematol. 2015, 102, 523–527. [Google Scholar] [CrossRef]
- Steinhilber, J.; Mederake, M.; Bonzheim, I.; Serinsoz-Linke, E.; Muller, I.; Fallier-Becker, P.; Lemonnier, F.; Gaulard, P.; Fend, F.; Quintanilla-Martinez, L. The pathological features of angioimmunoblastic T-cell lymphomas with IDH2(R172) mutations. Mod. Pathol. Off. J. USA Can. Acad. Pathol. Inc. 2019, 32, 1123–1134. [Google Scholar] [CrossRef]
- Nagao, R.; Kikuti, Y.Y.; Carreras, J.; Kikuchi, T.; Miyaoka, M.; Matsushita, H.; Kojima, M.; Ando, K.; Sakata-Yanagimoto, M.; Chiba, S.; et al. Clinicopathologic Analysis of Angioimmunoblastic T-cell Lymphoma with or without RHOA G17V Mutation Using Formalin-fixed Paraffin-embedded Sections. Am. J. Surg. Pathol. 2016, 40, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Ondrejka, S.L.; Grzywacz, B.; Bodo, J.; Makishima, H.; Polprasert, C.; Said, J.W.; Przychodzen, B.; Maciejewski, J.P.; Hsi, E.D. Angioimmunoblastic T-cell Lymphomas with the RHOA p.Gly17Val Mutation Have Classic Clinical and Pathologic Features. Am. J. Surg. Pathol. 2016, 40, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Moreau, A.; Dupuis, J.; Streubel, B.; Petit, B.; Le Gouill, S.; Martin-Garcia, N.; Copie-Bergman, C.; Gaillard, F.; Qubaja, M.; et al. Peripheral T-cell lymphomas with a follicular growth pattern are derived from follicular helper T cells (TFH) and may show overlapping features with angioimmunoblastic T-cell lymphomas. Am. J. Surg. Pathol. 2009, 33, 682–690. [Google Scholar] [CrossRef]
- Miyoshi, H.; Sato, K.; Niino, D.; Arakawa, F.; Kimura, Y.; Kiyasu, J.; Takeuchi, M.; Yoshida, M.; Okada, Y.; Nakamura, Y.; et al. Clinicopathologic analysis of peripheral T-cell lymphoma, follicular variant, and comparison with angioimmunoblastic T-cell lymphoma: Bcl-6 expression might affect progression between these disorders. Am. J. Clin. Pathol. 2012, 137, 879–889. [Google Scholar] [CrossRef] [PubMed]
- de Leval, L.; Savilo, E.; Longtine, J.; Ferry, J.A.; Harris, N.L. Peripheral T-cell lymphoma with follicular involvement and a CD4+/bcl-6+ phenotype. Am. J. Surg. Pathol. 2001, 25, 395–400. [Google Scholar] [CrossRef]
- Ikonomou, I.M.; Tierens, A.; Troen, G.; Aamot, H.V.; Heim, S.; Lauritzsen, G.F.; Valerhaugen, H.; Delabie, J. Peripheral T-cell lymphoma with involvement of the expanded mantle zone. Virchows Arch. Int. J. Pathol. 2006, 449, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, M.; Song, J.Y.; Sohani, A.R.; Moroch, J.; Plonquet, A.; Duffield, A.S.; Borowitz, M.J.; Jiang, L.; Bueso-Ramos, C.; Inamdar, K.; et al. Peripheral T-cell lymphomas of follicular helper T-cell type frequently display an aberrant CD3(-/dim)CD4(+) population by flow cytometry: An important clue to the diagnosis of a Hodgkin lymphoma mimic. Mod. Pathol. Off. J. USA Can. Acad. Pathol. Inc. 2016, 29, 1173–1182. [Google Scholar] [CrossRef]
- Dobay, M.P.; Lemonnier, F.; Missiaglia, E.; Bastard, C.; Vallois, D.; Jais, J.P.; Scourzic, L.; Dupuy, A.; Fataccioli, V.; Pujals, A.; et al. Integrative clinicopathological and molecular analyses of angioimmunoblastic T-cell lymphoma and other nodal lymphomas of follicular helper T-cell origin. Haematologica 2017, 102, e148–e151. [Google Scholar] [CrossRef]
- Debackere, K.; van der Krogt, J.A.; Tousseyn, T.; Ferreiro, J.A.F.; Van Roosbroeck, K.; Marcelis, L.; Graux, C.; Dierickx, D.; Ameye, G.; Vandenberghe, P.; et al. FER and FES tyrosine kinase fusions in follicular T-cell lymphoma. Blood 2020, 135, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Sakata-Yanagimoto, M.; Shimono, J.; Yoshida, N.; Hattori, K.; Arakawa, F.; Yanagida, E.; Takeuchi, M.; Yamada, K.; Suzuki, T.; et al. RHOA mutation in follicular T-cell lymphoma: Clinicopathological analysis of 16 cases. Pathol. Int. 2020, 70, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Streubel, B.; Vinatzer, U.; Willheim, M.; Raderer, M.; Chott, A. Novel t(5; 9)(q33; q22) fuses ITK to SYK in unspecified peripheral T-cell lymphoma. Leukemia 2006, 20, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Eladl, A.E.; Satou, A.; Elsayed, A.A.; Suzuki, Y.; Kato, S.; Asano, N.; Nakamura, S. Clinicopathological Study of 30 Cases of Peripheral T-cell Lymphoma with Hodgkin and Reed-Sternberg-like B-cells from Japan. Am. J. Surg. Pathol. 2017, 41, 506–516. [Google Scholar] [CrossRef]
- Nicolae, A.; Pittaluga, S.; Venkataraman, G.; Vijnovich-Baron, A.; Xi, L.; Raffeld, M.; Jaffe, E.S. Peripheral T-cell lymphomas of follicular T-helper cell derivation with Hodgkin/Reed-Sternberg cells of B-cell lineage: Both EBV-positive and EBV-negative variants exist. Am. J. Surg. Pathol. 2013, 37, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla-Martinez, L.; Fend, F.; Moguel, L.R.; Spilove, L.; Beaty, M.W.; Kingma, D.W.; Raffeld, M.; Jaffe, E.S. Peripheral T-cell lymphoma with Reed-Sternberg-like cells of B-cell phenotype and genotype associated with Epstein-Barr virus infection. Am. J. Surg. Pathol. 1999, 23, 1233–1240. [Google Scholar] [CrossRef]
- Sakakibara, A.; Kohno, K.; Eladl, A.E.; Klaisuwan, T.; Ishikawa, E.; Suzuki, Y.; Shimada, S.; Nakaguro, M.; Shimoyama, Y.; Takahara, T.; et al. Immunohistochemical assessment of the diagnostic utility of PD-L1: A preliminary analysis of anti-PD-L1 antibody (SP142) for lymphoproliferative diseases with tumour and non-malignant Hodgkin-Reed-Sternberg (HRS)-like cells. Histopathology 2018, 72, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Arisawa, K.; Soda, M.; Endo, S.; Kurokawa, K.; Katamine, S.; Shimokawa, I.; Koba, T.; Takahashi, T.; Saito, H.; Doi, H.; et al. Evaluation of adult T-cell leukemia/lymphoma incidence and its impact on non-Hodgkin lymphoma incidence in southwestern Japan. Int. J. Cancer 2000, 85, 319–324. [Google Scholar] [CrossRef]
- Tajima, K. The 4th nation-wide study of adult T-cell leukemia/lymphoma (ATL) in Japan: Estimates of risk of ATL and its geographical and clinical features. The T- and B-cell Malignancy Study Group. Int. J. Cancer 1990, 45, 237–243. [Google Scholar] [CrossRef]
- Tokudome, S.; Tokunaga, O.; Shimamoto, Y.; Miyamoto, Y.; Sumida, I.; Kikuchi, M.; Takeshita, M.; Ikeda, T.; Fujiwara, K.; Yoshihara, M.; et al. Incidence of adult T-cell leukemia/lymphoma among human T-lymphotropic virus type I carriers in Saga, Japan. Cancer Res. 1989, 49, 226–228. [Google Scholar] [PubMed]
- Matsuoka, M.; Jeang, K.T. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 2007, 7, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Shimoyama, M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984-87). Br. J. Haematol. 1991, 79, 428–437. [Google Scholar] [CrossRef]
- Ito, S.; Iwanaga, M.; Nosaka, K.; Imaizumi, Y.; Ishitsuka, K.; Amano, M.; Utsunomiya, A.; Tokura, Y.; Watanabe, T.; Uchimaru, K.; et al. Epidemiology of adult T-cell leukemia-lymphoma in Japan: An updated analysis, 2012–2013. Cancer Sci. 2021, 112, 4346–4354. [Google Scholar] [CrossRef] [PubMed]
- Katsuya, H.; Ishitsuka, K.; Utsunomiya, A.; Hanada, S.; Eto, T.; Moriuchi, Y.; Saburi, Y.; Miyahara, M.; Sueoka, E.; Uike, N.; et al. Treatment and survival among 1594 patients with ATL. Blood 2015, 126, 2570–2577. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, K.; Hermine, O.; Bazarbachi, A.; Ratner, L.; Ramos, J.C.; Harrington, W., Jr.; O’Mahony, D.; Janik, J.E.; Bittencourt, A.L.; Taylor, G.P.; et al. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: A proposal from an international consensus meeting. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 453–459. [Google Scholar] [CrossRef]
- Karube, K.; Suzumiya, J.; Okamoto, M.; Takeshita, M.; Maeda, K.; Sakaguchi, M.; Inada, T.; Tsushima, H.; Kikuchi, M.; Ohshima, K. Adult T-cell lymphoma/leukemia with angioimmunoblastic T-cell lymphomalike features: Report of 11 cases. Am. J. Surg. Pathol. 2007, 31, 216–223. [Google Scholar] [CrossRef]
- Ishida, T.; Utsunomiya, A.; Iida, S.; Inagaki, H.; Takatsuka, Y.; Kusumoto, S.; Takeuchi, G.; Shimizu, S.; Ito, M.; Komatsu, H.; et al. Clinical significance of CCR4 expression in adult T-cell leukemia/lymphoma: Its close association with skin involvement and unfavorable outcome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 3625–3634. [Google Scholar]
- Karube, K.; Ohshima, K.; Tsuchiya, T.; Yamaguchi, T.; Kawano, R.; Suzumiya, J.; Utsunomiya, A.; Harada, M.; Kikuchi, M. Expression of FoxP3, a key molecule in CD4CD25 regulatory T cells, in adult T-cell leukaemia/lymphoma cells. Br. J. Haematol. 2004, 126, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Karube, K.; Aoki, R.; Sugita, Y.; Yoshida, S.; Nomura, Y.; Shimizu, K.; Kimura, Y.; Hashikawa, K.; Takeshita, M.; Suzumiya, J.; et al. The relationship of FOXP3 expression and clinicopathological characteristics in adult T-cell leukemia/lymphoma. Mod. Pathol. Off. J. USA Can. Acad. Pathol. Inc. 2008, 21, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Suzumiya, J.; Kato, A.; Tashiro, K.; Kikuchi, M. Clonal HTLV-I-infected CD4+ T-lymphocytes and non-clonal non-HTLV-I-infected giant cells in incipient ATLL with Hodgkin-like histologic features. Int. J. Cancer 1997, 72, 592–598. [Google Scholar] [CrossRef]
- Venkataraman, G.; Berkowitz, J.; Morris, J.C.; Janik, J.E.; Raffeld, M.A.; Pittaluga, S. Adult T-cell leukemia/lymphoma with Epstein-Barr virus-positive Hodgkin-like cells. Hum. Pathol. 2011, 42, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Karube, K.; Takatori, M.; Sakihama, S.; Tsuruta, Y.; Miyagi, T.; Morichika, K.; Kitamura, S.; Nakada, N.; Hayashi, M.; Tomori, S.; et al. Clinicopathological features of adult T-cell leukemia/lymphoma with HTLV-1-infected Hodgkin and Reed-Sternberg-like cells. Blood Adv. 2021, 5, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Shah, U.A.; Chung, E.Y.; Giricz, O.; Pradhan, K.; Kataoka, K.; Gordon-Mitchell, S.; Bhagat, T.D.; Mai, Y.; Wei, Y.; Ishida, E.; et al. North American ATLL has a distinct mutational and transcriptional profile and responds to epigenetic therapies. Blood 2018, 132, 1507–1518. [Google Scholar] [CrossRef]
- Kogure, Y.; Kataoka, K. Genetic alterations in adult T-cell leukemia/lymphoma. Cancer Sci. 2017, 108, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Miyoshi, H.; Ohshima, K. Clinical Applications of Genomic Alterations in ATLL: Predictive Markers and Therapeutic Targets. Cancers 2021, 13, 1801. [Google Scholar] [CrossRef]
- Yoshida, N.; Shigemori, K.; Donaldson, N.; Trevisani, C.; Cordero, N.A.; Stevenson, K.E.; Bu, X.; Arakawa, F.; Takeuchi, M.; Ohshima, K.; et al. Genomic landscape of young ATLL patients identifies frequent targetable CD28 fusions. Blood 2020, 135, 1467–1471. [Google Scholar] [CrossRef]
- Kataoka, K.; Shiraishi, Y.; Takeda, Y.; Sakata, S.; Matsumoto, M.; Nagano, S.; Maeda, T.; Nagata, Y.; Kitanaka, A.; Mizuno, S.; et al. Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature 2016, 534, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Karube, K.; Utsunomiya, A.; Tsukasaki, K.; Imaizumi, Y.; Taira, N.; Uike, N.; Umino, A.; Arita, K.; Suguro, M.; et al. Molecular characterization of chronic-type adult T-cell leukemia/lymphoma. Cancer Res. 2014, 74, 6129–6138. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Kiyasu, J.; Kato, T.; Yoshida, N.; Shimono, J.; Yokoyama, S.; Taniguchi, H.; Sasaki, Y.; Kurita, D.; Kawamoto, K.; et al. PD-L1 expression on neoplastic or stromal cells is respectively a poor or good prognostic factor for adult T-cell leukemia/lymphoma. Blood 2016, 128, 1374–1381. [Google Scholar] [CrossRef]
- Takeuchi, M.; Miyoshi, H.; Asano, N.; Yoshida, N.; Yamada, K.; Yanagida, E.; Moritsubo, M.; Nakata, M.; Umeno, T.; Suzuki, T.; et al. Human leukocyte antigen class II expression is a good prognostic factor in adult T-cell leukemia/lymphoma. Haematologica 2019, 104, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Miyoshi, H.; Kato, T.; Sakata-Yanagimoto, M.; Niino, D.; Taniguchi, H.; Moriuchi, Y.; Miyahara, M.; Kurita, D.; Sasaki, Y.; et al. CCR4 frameshift mutation identifies a distinct group of adult T cell leukaemia/lymphoma with poor prognosis. J. Pathol. 2016, 238, 621–626. [Google Scholar] [CrossRef]
- Nakagawa, M.; Schmitz, R.; Xiao, W.; Goldman, C.K.; Xu, W.; Yang, Y.; Yu, X.; Waldmann, T.A.; Staudt, L.M. Gain-of-function CCR4 mutations in adult T cell leukemia/lymphoma. J. Exp. Med. 2014, 211, 2497–2505. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Ishida, T.; Masaki, A.; Murase, T.; Yonekura, K.; Tashiro, Y.; Tokunaga, M.; Utsunomiya, A.; Ito, A.; Kusumoto, S.; et al. CCR4 mutations associated with superior outcome of adult T-cell leukemia/lymphoma under mogamulizumab treatment. Blood 2018, 132, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Iwanaga, M.; Yasunaga, J.I.; Nagata, Y.; Kitanaka, A.; Kameda, T.; Yoshimitsu, M.; Shiraishi, Y.; Sato-Otsubo, A.; Sanada, M.; et al. Prognostic relevance of integrated genetic profiling in adult T-cell leukemia/lymphoma. Blood 2018, 131, 215–225. [Google Scholar] [CrossRef]
- Yoshida, N.; Weinstock, D.M. Clinicogenetic risk modeling in ATL. Blood 2018, 131, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Weisenburger, D.D.; Savage, K.J.; Harris, N.L.; Gascoyne, R.D.; Jaffe, E.S.; MacLennan, K.A.; Rudiger, T.; Pileri, S.; Nakamura, S.; Nathwani, B.; et al. Peripheral T-cell lymphoma, not otherwise specified: A report of 340 cases from the International Peripheral T-cell Lymphoma Project. Blood 2011, 117, 3402–3408. [Google Scholar] [CrossRef] [PubMed]
- Geissinger, E.; Odenwald, T.; Lee, S.S.; Bonzheim, I.; Roth, S.; Reimer, P.; Wilhelm, M.; Muller-Hermelink, H.K.; Rudiger, T. Nodal peripheral T-cell lymphomas and, in particular, their lymphoepithelioid (Lennert’s) variant are often derived from CD8(+) cytotoxic T-cells. Virchows Arch. Int. J. Pathol. 2004, 445, 334–343. [Google Scholar] [CrossRef]
- Yamashita, Y.; Nakamura, S.; Kagami, Y.; Hasegawa, Y.; Kojima, H.; Nagasawa, T.; Mori, N. Lennert’s lymphoma: A variant of cytotoxic T-cell lymphoma? Am. J. Surg. Pathol. 2000, 24, 1627–1633. [Google Scholar] [CrossRef]
- Spier, C.M.; Lippman, S.M.; Miller, T.P.; Grogan, T.M. Lennert’s lymphoma. A clinicopathologic study with emphasis on phenotype and its relationship to survival. Cancer 1988, 61, 517–524. [Google Scholar] [CrossRef]
- Went, P.; Agostinelli, C.; Gallamini, A.; Piccaluga, P.P.; Ascani, S.; Sabattini, E.; Bacci, F.; Falini, B.; Motta, T.; Paulli, M.; et al. Marker expression in peripheral T-cell lymphoma: A proposed clinical-pathologic prognostic score. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 2472–2479. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Suzuki, R.; Kagami, Y.; Ishida, F.; Kitamura, K.; Fukutani, H.; Morishima, Y.; Takeuchi, K.; Nakamura, S. Clinicopathologic and prognostic significance of cytotoxic molecule expression in nodal peripheral T-cell lymphoma, unspecified. Am. J. Surg. Pathol. 2005, 29, 1284–1293. [Google Scholar] [CrossRef]
- Yamashita, D.; Shimada, K.; Takata, K.; Miyata-Takata, T.; Kohno, K.; Satou, A.; Sakakibara, A.; Nakamura, S.; Asano, N.; Kato, S. Reappraisal of nodal Epstein-Barr Virus-negative cytotoxic T-cell lymphoma: Identification of indolent CD5(+) diseases. Cancer Sci. 2018, 109, 2599–2610. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Takahashi, E.; Asano, N.; Tanaka, T.; Megahed, N.; Kinoshita, T.; Nakamura, S. Nodal cytotoxic molecule (CM)-positive Epstein-Barr virus (EBV)-associated peripheral T cell lymphoma (PTCL): A clinicopathological study of 26 cases. Histopathology 2012, 61, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Asano, N.; Miyata-Takata, T.; Takata, K.; Elsayed, A.A.; Satou, A.; Takahashi, E.; Kinoshita, T.; Nakamura, S. T-cell receptor (TCR) phenotype of nodal Epstein-Barr virus (EBV)-positive cytotoxic T-cell lymphoma (CTL): A clinicopathologic study of 39 cases. Am. J. Surg. Pathol. 2015, 39, 462–471. [Google Scholar] [CrossRef]
- Kato, S.; Yamashita, D.; Nakamura, S. Nodal EBV+ cytotoxic T-cell lymphoma: A literature review based on the 2017 WHO classification. J. Clin. Exp. Hematop. 2020, 60, 30–36. [Google Scholar] [CrossRef]
- Iqbal, J.; Wright, G.; Wang, C.; Rosenwald, A.; Gascoyne, R.D.; Weisenburger, D.D.; Greiner, T.C.; Smith, L.; Guo, S.; Wilcox, R.A.; et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood 2014, 123, 2915–2923. [Google Scholar] [CrossRef]
- Heavican, T.B.; Bouska, A.; Yu, J.; Lone, W.; Amador, C.; Gong, Q.; Zhang, W.; Li, Y.; Dave, B.J.; Nairismagi, M.L.; et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood 2019, 133, 1664–1676. [Google Scholar] [CrossRef]
- Amador, C.; Greiner, T.C.; Heavican, T.B.; Smith, L.M.; Galvis, K.T.; Lone, W.; Bouska, A.; D’Amore, F.; Pedersen, M.B.; Pileri, S.; et al. Reproducing the molecular subclassification of peripheral T-cell lymphoma-NOS by immunohistochemistry. Blood 2019, 134, 2159–2170. [Google Scholar] [CrossRef]
- Watatani, Y.; Sato, Y.; Miyoshi, H.; Sakamoto, K.; Nishida, K.; Gion, Y.; Nagata, Y.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; et al. Molecular heterogeneity in peripheral T-cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia 2019, 33, 2867–2883. [Google Scholar] [CrossRef] [PubMed]
- Maura, F.; Dodero, A.; Carniti, C.; Bolli, N.; Magni, M.; Monti, V.; Cabras, A.; Leongamornlert, D.; Abascal, F.; Diamond, B.; et al. CDKN2A deletion is a frequent event associated with poor outcome in patients with peripheral T-cell lymphoma not otherwise specified (PTCL-NOS). Haematologica 2021, 106, 2918–2926. [Google Scholar] [CrossRef] [PubMed]
- Laginestra, M.A.; Cascione, L.; Motta, G.; Fuligni, F.; Agostinelli, C.; Rossi, M.; Sapienza, M.R.; Righi, S.; Broccoli, A.; Indio, V.; et al. Whole exome sequencing reveals mutations in FAT1 tumor suppressor gene clinically impacting on peripheral T-cell lymphoma not otherwise specified. Mod. Pathol. Off. J. USA Can. Acad. Pathol. Inc. 2020, 33, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Sugio, T.; Miyawaki, K.; Kato, K.; Sasaki, K.; Yamada, K.; Iqbal, J.; Miyamoto, T.; Ohshima, K.; Maeda, T.; Miyoshi, H.; et al. Microenvironmental immune cell signatures dictate clinical outcomes for PTCL-NOS. Blood Adv. 2018, 2, 2242–2252. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Translocation | Partner Gene | ALK Staining Pattern | Frequency |
|---|---|---|---|
| t(2;5)(p23;q35) | NPM1 | Cytoplasmic and nuclear | 80% |
| t(1;2)(q25;p23) | TPM3 | Cytoplasmic with peripheral intensification | 10–15% |
| inv(2)(p23q35) | ATIC | Cytoplasmic | 1% |
| t(2;3)(p23;q12.2) | TFG | Cytoplasmic | <1% |
| t(2;17)(p23;q23) | CLTC | Cytoplasmic | <1% |
| t(2;22)(p23;q11.2) | MYH9 | Cytoplasmic | <1% |
| t(2;17)(p23;q23) | RNF213 | Cytoplasmic | <1% |
| t(X;2)(q11-12;p23) | MSN | Membrane | <1% |
| t(2;9)(p23;p13.1) | TPM4 | Cytoplasmic | <1% |
| t(2;9)(p23;q33) | TRAF1 | Cytoplasmic | <1% |
| t(2;11)(p23;qR3) | EEFIG | Cytoplasmic | <1% |
| t(2;8)(p23;q22) | PABCP1 | Cytoplasmic | <1% |
| Genetic Alterations and Biomarkers | Subtype | Notes |
|---|---|---|
| CD30 expression | ALCL | Expression in 100% (by definition); target of BV |
| ALK rearrangement | ALK+ ALCL | Translocations in 100% (by definition); sensitive to ALK inhibitors |
| DUSP22 rearrangement [21] | ALK− ALCL | Translocation in 30%; favorable prognosis |
| TP63 rearrangement [21] | ALK− ALCL | Translocation in 8%; dismal prognosis |
| JAK1 and STAT3 [29] | ALK− ALCL | JAK1 mutations in 15% and STAT3 mutations in ~10%; activation of the JAK-STAT3 pathway |
| Genetic Alterations and Biomarkers | Subtype | Notes |
|---|---|---|
| TET2 mutation [43,44,45,62] | TFH lymphoma | Loss of function mutations in 40–80% of AITL; slightly more frequent in FTCL and nPTCL-TFH |
| DNMT3A mutation [43,44,45,62] | TFH lymphoma | Loss of function mutations in 30% of AITL |
| IDH2R172 mutation [46,47,62] | AITL and FTCL | Mutations are restricted to AITL (30%) and FTCL; increased DNA methylation |
| RHOAG17V mutation [44,48,49,50,62,64] | TFH lymphoma | Mutations in 50–71% of AITL; loss of RHOA GTPase activity |
| ITK-SYK fusion [65] | FTCL | Fusions in 20%; specific for FTCL; sensitivity to the SYC inhibitor |
| Genetic Alterations and Biomarkers | Subtype | Notes |
|---|---|---|
| Genomic alterations affecting the TCR/NF-κβ pathway [85,86,87,88] | ATLL | Genetic alteration in up to 90%; genetic alterations of a proximal component of TCR signaling (e.g., FYN, PLCG1, and VAV1), of the NF-κβ pathway (e.g., PRKCB and CARD11), and of downstream signaling (e.g., IRF4 and RHOA). |
| Fusions related to CD28 [89] | ATLL | In-frame fusions in 10%; CTLA4-CD28 and ICOS-CD28; contribute to NF-κβ pathway activation |
| PD-L1 amplification [85] | ATLL | Copy number gains in 15%; potential target of the immune checkpoint inhibitor; worse prognosis |
| PD-L1 disruption of the 3′-UTR [90] | ATLL | Disruption of the 3’-UTR in 27%; potential target of the immune checkpoint inhibitor |
| PD-L1 expression [92] | ATLL | Expression in 7.4%; worse prognosis |
| CCR4 mutation [85,94,95] | ATLL | Mutations in 30%; up-regulation of CCR4 expression |
| CCR4 expression [79] | ATLL | Expression in 90%; worse prognosis; good candidate for the anti-CCR4 antibody (mogamulizumab) |
| Genetic Alterations and Biomarkers | Subtype | Notes |
|---|---|---|
| GATA3 and/or CCR expression [111] | GATA3 subgroup | Biological subgroup of PTCL-NOS; 30–40% of PTCL-NOS; worse prognosis |
| TBX21/CXCR3 expression [111] | TBX21 subgroup | Biological subgroup of PTCL-NOS; 50–60% of PTCL-NOS; better prognosis than that of the GATA3 subgroup |
| TP53 mutation and deletion [110,112] | PTCL-NOS | Frequent in the GATA-3 subgroup; worse prognosis |
| CDKN2A deletion [110,112,113] | PTCL-NOS | Frequent in the GATA-3 subgroup; worse prognosis |
| PTEN deletion [110,112,113] | PTCL-NOS | Frequent in the GATA-3 subgroup; worse prognosis |
| FAT1 mutation [114] | PTCL-NOS | Mutations in 39%; worse prognosis |
| CM expression [100,103,104] | nPTCL-NOS | Expression in 15–40%; aggressive clinical behavior; worse prognosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Satou, A.; Takahara, T.; Tsuzuki, T. Pathological and Molecular Features of Nodal Peripheral T-Cell Lymphomas. Diagnostics 2022, 12, 2001. https://doi.org/10.3390/diagnostics12082001
Satou A, Takahara T, Tsuzuki T. Pathological and Molecular Features of Nodal Peripheral T-Cell Lymphomas. Diagnostics. 2022; 12(8):2001. https://doi.org/10.3390/diagnostics12082001
Chicago/Turabian StyleSatou, Akira, Taishi Takahara, and Toyonori Tsuzuki. 2022. "Pathological and Molecular Features of Nodal Peripheral T-Cell Lymphomas" Diagnostics 12, no. 8: 2001. https://doi.org/10.3390/diagnostics12082001
APA StyleSatou, A., Takahara, T., & Tsuzuki, T. (2022). Pathological and Molecular Features of Nodal Peripheral T-Cell Lymphomas. Diagnostics, 12(8), 2001. https://doi.org/10.3390/diagnostics12082001