
The Value of Electroretinography in Identifying Candidate Genes for Inherited Retinal Dystrophies: A Diagnostic Guide

 ,
,  ,
, 
Abstract
:1. Introduction
1.1. Background and Significance of Electroretinography (ERG)
1.2. Anatomy and Physiology of the Retina
1.3. Principles of ERG Recording and Methodological Details
1.4. ERG Waveforms and Components
1.5. Electronegative ERG: Pathophysiology and Associated Diseases
2. Congenital Stationary Night Blindness (CSNB): Riggs, Complete (cCSNB), and Incomplete (iCSNB)
2.1. Pathophysiology
2.2. Clinical Presentation
2.3. ERG Findings and Candidate Genes
2.4. Differential Diagnosis, Treatments, and Prognosis
3. X-Linked Retinoschisis (XLRS)
3.1. Pathophysiology
3.2. Clinical Presentation
3.3. ERG Findings and Diagnostic Criteria
3.4. Management and Prognosis
4. CRX-Related Cone–Rod Dystrophy
4.1. Pathophysiology
4.2. Clinical Presentation
4.3. ERG Findings
4.4. Other Genes with These Features
5. Fundus Albipunctatus
5.1. Pathophysiology
5.2. Clinical Presentation
5.3. ERG Findings
5.4. Differential Diagnosis
6. Enhanced S-Cone Syndrome (ESCS), or Goldmann–Favre Syndrome
6.1. Pathophysiology
6.2. Clinical Presentation
6.3. ERG Findings
6.4. Differential Diagnosis
7. Cone Dystrophy with Supernormal Rod Response (CDSRR)
7.1. Pathophysiology
7.2. Clinical Presentation
7.3. ERG Findings
7.4. Differential Diagnosis
8. Challenges and Limitations of ERG in Diagnosis
8.1. Technical Considerations
8.2. Variability and Standardization of ERG Protocols
8.3. Importance of Integrating Clinical Findings and Multimodal Imaging with ERG
9. Conclusions
9.1. Summary of Findings
9.2. Clinical Implications and the Unique Role of ERG in Diagnosing Select Retinal Disorders
9.3. Limitations and Future Research Advancements in ERG Applications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Human Genome Research Institute. The Cost of Sequencing a Human Genome. Available online: https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost (accessed on 28 May 2023).
- Vickers, A.J.; Van Calster, B.; Steyerberg, E.W. Net benefit approaches to the evaluation of prediction models, molecular markers, and diagnostic tests. BMJ 2016, 352, i6. [Google Scholar] [CrossRef] [PubMed]
- Bertozzi, S.; Londero, A.P.; Xholli, A.; Azioni, G.; Di Vora, R.; Paudice, M.; Bucimazza, I.; Cedolini, C.; Cagnacci, A. Risk-Reducing Breast and Gynecological Surgery for BRCA Mutation Carriers: A Narrative Review. J. Clin. Med. 2023, 12, 1422. [Google Scholar] [CrossRef] [PubMed]
- Purves, D.; Augustine, G.J.; Fitzpatrick, D. Neuroscience. In Anatomical Distribution of Rods and Cones, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2001. [Google Scholar]
- Lamb, T.D. Photoreceptor physiology and evolution: Cellular and molecular basis of rod and cone phototransduction. J. Physiol. 2022, 600, 4585–4601. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, T.; Habib, S. ON and OFF Signaling Pathways in the Retina and the Visual System. Front. Ophthalmol. 2022, 2, 989002. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.G.; Frishman, L.J.; Grigg, J.; Hamilton, R.; Jeffrey, B.G.; Kondo, M.; Li, S.; McCulloch, D.L. ISCEV Standard for full-field clinical electroretinography (2022 update). Doc. Ophthalmol. 2022, 144, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Robson, A.G.; Holder, G.E.; Moore, A.T. The negative ERG: Clinical phenotypes and disease mechanisms of inner retinal dysfunction. Surv. Ophthalmol. 2008, 53, 16–40. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Mahroo, O.A. Negative electroretinograms: Genetic and acquired causes, diagnostic approaches and physiological insights. Eye 2021, 35, 2419–2437. [Google Scholar] [CrossRef] [PubMed]
- Vercio, A.; Chalam, K.V.; Winter, T. Typical electronegative electroretinography and nyctalopia as a presenting feature of systemic malignant melanoma. Int. Med. Case Rep. J. 2019, 12, 265–276. [Google Scholar] [CrossRef]
- Chiang, T.K.; White, K.M.; Kurup, S.K.; Yu, M. Use of Visual Electrophysiology to Monitor Retinal and Optic Nerve Toxicity. Biomolecules 2022, 12, 1390. [Google Scholar] [CrossRef]
- Kim, H.M.; Park, K.H.; Woo, S.J. Correlation of electroretinography components with visual function and prognosis of central retinal artery occlusion. Sci. Rep. 2020, 10, 12146. [Google Scholar] [CrossRef]
- Saker, S.; Morales, M.; Jhittay, H.; Wen, Y.; Amoaku, W. Electrophysiological and microperimetry changes in vitamin A deficiency retinopathy. Doc. Ophthalmol. 2015, 130, 231–240. [Google Scholar] [CrossRef]
- Tsang, S.H.; Sharma, T. Congenital Stationary Night Blindness. Adv. Exp. Med. Biol. 2018, 1085, 61–64. [Google Scholar] [CrossRef] [PubMed]
- De Silva, S.R.; Arno, G.; Robson, A.G.; Fakin, A.; Pontikos, N.; Mohamed, M.D.; Bird, A.C.; Moore, A.T.; Michaelides, M.; Webster, A.R.; et al. The X-linked retinopathies: Physiological insights, pathogenic mechanisms, phenotypic features and novel therapies. Prog. Retin. Eye Res. 2021, 82, 100898. [Google Scholar] [CrossRef] [PubMed]
- Morgans, C.W.; Brown, R.L.; Duvoisin, R.M. TRPM1: The endpoint of the mGluR6 signal transduction cascade in retinal ON-bipolar cells. Bioessays 2010, 32, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Morgans, C.W.; Zhang, J.; Jeffrey, B.G.; Nelson, S.M.; Burke, N.S.; Duvoisin, R.M.; Brown, R.L. TRPM1 is required for the depolarizing light response in retinal ON-bipolar cells. Proc. Natl. Acad. Sci. USA 2009, 106, 19174–19178. [Google Scholar] [CrossRef] [PubMed]
- Schneider, F.M.; Mohr, F.; Behrendt, M.; Oberwinkler, J. Properties and functions of TRPM1 channels in the dendritic tips of retinal ON-bipolar cells. Eur. J. Cell Biol. 2015, 94, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Pearring, J.N.; Bojang, P., Jr.; Shen, Y.; Koike, C.; Furukawa, T.; Nawy, S.; Gregg, R.G. A role for nyctalopin, a small leucine-rich repeat protein, in localizing the TRP melastatin 1 channel to retinal depolarizing bipolar cell dendrites. J. Neurosci. 2011, 31, 10060–10066. [Google Scholar] [CrossRef]
- Hasan, N.; Pangeni, G.; Cobb, C.A.; Ray, T.A.; Nettesheim, E.R.; Ertel, K.J.; Lipinski, D.M.; McCall, M.A.; Gregg, R.G. Presynaptic Expression of LRIT3 Transsynaptically Organizes the Postsynaptic Glutamate Signaling Complex Containing TRPM1. Cell Rep. 2019, 27, 3107–3116.e3103. [Google Scholar] [CrossRef]
- Kim, A.H.; Liu, P.K.; Chang, Y.H.; Kang, E.Y.; Wang, H.H.; Chen, N.; Tseng, Y.J.; Seo, G.H.; Lee, H.; Liu, L.; et al. Congenital Stationary Night Blindness: Clinical and Genetic Features. Int. J. Mol. Sci. 2022, 23, 14965. [Google Scholar] [CrossRef]
- Gravier, N. [Etiological assessment of a nystagmus in childhood]. J. Fr. Ophtalmol. 2018, 41, 868–878. [Google Scholar] [CrossRef]
- Blair, K.; Cibis, G.; Gulani, A.C. Amblyopia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://pubmed.ncbi.nlm.nih.gov/28613640/ (accessed on 1 August 2023).
- MacDonald, I.M.; Hoang, S.; Tuupanen, S. X-Linked Congenital Stationary Night Blindness. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Kim, H.M.; Joo, K.; Han, J.; Woo, S.J. Clinical and Genetic Characteristics of Korean Congenital Stationary Night Blindness Patients. Genes 2021, 12, 789. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.F.; Zeitz, C. Riggs-type dominant congenital stationary night blindness: ERG findings, a new GNAT1 mutation and a systemic association. Doc. Ophthalmol. 2018, 137, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Hung, W.C.; Cheng, H.C.; Wang, A.G. Melanoma-associated retinopathy with anti-TRPM1 autoantibodies showing concomitant Off-bipolar cell dysfunction. Doc. Ophthalmol. 2022, 145, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Liu, P.K.; Wang, N.K. Electroretinogram (ERG) to Evaluate the Retina in Cases of Retinitis Pigmentosa (RP). Methods Mol. Biol. 2023, 2560, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Dryja, T.P.; McGee, T.L.; Berson, E.L.; Fishman, G.A.; Sandberg, M.A.; Alexander, K.R.; Derlacki, D.J.; Rajagopalan, A.S. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc. Natl. Acad. Sci. USA 2005, 102, 4884–4889. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Ito, S.; Piao, C.H.; Terasaki, H.; Miyake, Y. Retinal and optic disc atrophy associated with a CACNA1F mutation in a Japanese family. Arch. Ophthalmol. 2003, 121, 1028–1033. [Google Scholar] [CrossRef]
- Scalabrino, M.L.; Boye, S.L.; Fransen, K.M.; Noel, J.M.; Dyka, F.M.; Min, S.H.; Ruan, Q.; De Leeuw, C.N.; Simpson, E.M.; Gregg, R.G.; et al. Intravitreal delivery of a novel AAV vector targets ON bipolar cells and restores visual function in a mouse model of complete congenital stationary night blindness. Hum. Mol. Genet. 2015, 24, 6229–6239. [Google Scholar] [CrossRef]
- Varin, J.; Bouzidi, N.; Dias, M.M.S.; Pugliese, T.; Michiels, C.; Robert, C.; Desrosiers, M.; Sahel, J.A.; Audo, I.; Dalkara, D.; et al. Restoration of mGluR6 Localization Following AAV-Mediated Delivery in a Mouse Model of Congenital Stationary Night Blindness. Investig. Ophthalmol. Vis. Sci. 2021, 62, 24. [Google Scholar] [CrossRef]
- Varin, J.; Bouzidi, N.; Gauvain, G.; Joffrois, C.; Desrosiers, M.; Robert, C.; De Sousa Dias, M.M.; Neuillé, M.; Michiels, C.; Nassisi, M.; et al. Substantial restoration of night vision in adult mice with congenital stationary night blindness. Mol. Ther. Methods Clin. Dev. 2021, 22, 15–25. [Google Scholar] [CrossRef]
- Miyadera, K.; Santana, E.; Roszak, K.; Iffrig, S.; Visel, M.; Iwabe, S.; Boyd, R.F.; Bartoe, J.T.; Sato, Y.; Gray, A.; et al. Targeting ON-bipolar cells by AAV gene therapy stably reverses LRIT3-congenital stationary night blindness. Proc. Natl. Acad. Sci. USA 2022, 119, e2117038119. [Google Scholar] [CrossRef]
- Chiu, W.; Lin, T.Y.; Chang, Y.C.; Isahwan-Ahmad Mulyadi Lai, H.; Lin, S.C.; Ma, C.; Yarmishyn, A.A.; Lin, S.C.; Chang, K.J.; Chou, Y.B.; et al. An Update on Gene Therapy for Inherited Retinal Dystrophy: Experience in Leber Congenital Amaurosis Clinical Trials. Int. J. Mol. Sci. 2021, 22, 4534. [Google Scholar] [CrossRef] [PubMed]
- Holmes, J.M.; Lazar, E.L.; Melia, B.M.; Astle, W.F.; Dagi, L.R.; Donahue, S.P.; Frazier, M.G.; Hertle, R.W.; Repka, M.X.; Quinn, G.E.; et al. Effect of age on response to amblyopia treatment in children. Arch. Ophthalmol. 2011, 129, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Molday, R.S.; Kellner, U.; Weber, B.H. X-linked juvenile retinoschisis: Clinical diagnosis, genetic analysis, and molecular mechanisms. Prog. Retin. Eye Res. 2012, 31, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Orès, R.; Mohand-Said, S.; Dhaenens, C.M.; Antonio, A.; Zeitz, C.; Augstburger, E.; Andrieu, C.; Sahel, J.A.; Audo, I. Phenotypic Characteristics of a French Cohort of Patients with X-Linked Retinoschisis. Ophthalmology 2018, 125, 1587–1596. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Sun, W.; Xiao, X.; Li, S.; Luo, H.; Jia, X.; Ouyang, J.; Li, X.; Wang, Y.; Jiang, Y.; et al. Clinical and genetic features of retinoschisis in 120 families with RS1 mutations. Br. J. Ophthalmol. 2023, 107, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.K.; Liu, L.; Chen, H.M.; Tsai, S.; Chang, T.C.; Tsai, T.H.; Yang, C.M.; Chao, A.N.; Chen, K.J.; Kao, L.Y.; et al. Clinical presentations of X-linked retinoschisis in Taiwanese patients confirmed with genetic sequencing. Mol. Vis. 2015, 21, 487–501. [Google Scholar] [PubMed]
- Parra, M.M.; Hartnett, M.E. Vitreous hemorrhage in X-linked retinoschisis. Am. J. Ophthalmol. Case Rep. 2022, 25, 101395. [Google Scholar] [CrossRef]
- Greig, L.C.; Gutierrez, K.G.; Oh, J.K.; Levi, S.R.; Korot, E.; Tsang, S.H.; Mahajan, V.B. Multimodal imaging reveals retinoschisis masquerading as retinal detachment in patients with choroideremia. Am. J. Ophthalmol. Case Rep. 2022, 26, 101543. [Google Scholar] [CrossRef]
- Tondelli, N.N.; Mencaroni, B.M.; Lemos, C.M.B.; Rocha de Sousa, J.; Sandoval Barbosa, G.C.; Gomes, A.M.V.; da Palma, M.M. A patient with X-linked retinoschisis and exudative retinal detachment associated with a pathogenic hemizygous variant c.304c>T in RS1. Ophthalmic Genet. 2022, 43, 871–875. [Google Scholar] [CrossRef]
- Wolfensberger, T.J.; Mahieu, I.; Jarvis-Evans, J.; Boulton, M.; Carter, N.D.; Nógrádi, A.; Hollande, E.; Bird, A.C. Membrane-bound carbonic anhydrase in human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 1994, 35, 3401–3407. [Google Scholar]
- Wolfensberger, T.J. The role of carbonic anhydrase inhibitors in the management of macular edema. Doc. Ophthalmol. 1999, 97, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.A.; Wang, K.; DeBenedictis, M.J.; Traboulsi, E.I. Topical carbonic anhydrase inhibitors in the long-term treatment of juvenile X-linked retinoschisis. Retina 2022, 42, 2176–2183. [Google Scholar] [CrossRef] [PubMed]
- Khandhadia, S.; Trump, D.; Menon, G.; Lotery, A.J. X-linked retinoschisis maculopathy treated with topical dorzolamide, and relationship to genotype. Eye 2011, 25, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Bucher, K.; Rodríguez-Bocanegra, E.; Dauletbekov, D.; Fischer, M.D. Immune responses to retinal gene therapy using adeno-associated viral vectors—Implications for treatment success and safety. Prog. Retin. Eye Res. 2021, 83, 100915. [Google Scholar] [CrossRef] [PubMed]
- Cukras, C.; Wiley, H.E.; Jeffrey, B.G.; Sen, H.N.; Turriff, A.; Zeng, Y.; Vijayasarathy, C.; Marangoni, D.; Ziccardi, L.; Kjellstrom, S.; et al. Retinal AAV8-RS1 Gene Therapy for X-Linked Retinoschisis: Initial Findings from a Phase I/IIa Trial by Intravitreal Delivery. Mol. Ther. 2018, 26, 2282–2294. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Y.; Punzo, C. Update on Viral Gene Therapy Clinical Trials for Retinal Diseases. Hum. Gene Ther. 2022, 33, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Hutton, D. Atsena Therapeutics Receives FDA Clearance of IND Application for ATSN-201 Investigational Gene Therapy for Treatment of X-Linked Retinoschisis. Available online: https://atsenatx.com/press-release/atsena-therapeutics-receives-fda-clearance-of-ind-application-for-atsn-201-an-investigational-gene-therapy-for-the-treatment-of-x-linked-retinoschisis/ (accessed on 1 May 2023).
- Furukawa, T.; Morrow, E.M.; Cepko, C.L. Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell 1997, 91, 531–541. [Google Scholar] [CrossRef]
- Freund, C.L.; Gregory-Evans, C.Y.; Furukawa, T.; Papaioannou, M.; Looser, J.; Ploder, L.; Bellingham, J.; Ng, D.; Herbrick, J.A.; Duncan, A.; et al. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 1997, 91, 543–553. [Google Scholar] [CrossRef]
- Lines, M.A.; Hébert, M.; McTaggart, K.E.; Flynn, S.J.; Tennant, M.T.; MacDonald, I.M. Electrophysiologic and phenotypic features of an autosomal cone-rod dystrophy caused by a novel CRX mutation. Ophthalmology 2002, 109, 1862–1870. [Google Scholar] [CrossRef]
- Molday, R.S.; Hicks, D.; Molday, L. Peripherin. A rim-specific membrane protein of rod outer segment discs. Investig. Ophthalmol. Vis. Sci. 1987, 28, 50–61. [Google Scholar]
- Arikawa, K.; Molday, L.L.; Molday, R.S.; Williams, D.S. Localization of peripherin/rds in the disk membranes of cone and rod photoreceptors: Relationship to disk membrane morphogenesis and retinal degeneration. J. Cell Biol. 1992, 116, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Nishiguchi, K.M.; Kunikata, H.; Fujita, K.; Hashimoto, K.; Koyanagi, Y.; Akiyama, M.; Ikeda, Y.; Momozawa, Y.; Sonoda, K.H.; Murakami, A.; et al. Association of CRX genotypes and retinal phenotypes confounded by variable expressivity and electronegative electroretinogram. Clin. Exp. Ophthalmol. 2020, 48, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Chapi, M.; Sabbaghi, H.; Suri, F.; Alehabib, E.; Rahimi-Aliabadi, S.; Jamali, F.; Jamshidi, J.; Emamalizadeh, B.; Darvish, H.; Mirrahimi, M.; et al. Incomplete penetrance of CRX gene for autosomal dominant form of cone-rod dystrophy. Ophthalmic Genet. 2019, 40, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.L.; Chen, S.; Esumi, N.; Swain, P.K.; Haines, H.S.; Peng, G.; Melia, B.M.; McIntosh, I.; Heckenlively, J.R.; Jacobson, S.G.; et al. QRX, a novel homeobox gene, modulates photoreceptor gene expression. Hum. Mol. Genet. 2004, 13, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Gregory-Evans, K.; Kelsell, R.E.; Gregory-Evans, C.Y.; Downes, S.M.; Fitzke, F.W.; Holder, G.E.; Simunovic, M.; Mollon, J.D.; Taylor, R.; Hunt, D.M.; et al. Autosomal dominant cone-rod retinal dystrophy (CORD6) from heterozygous mutation of GUCY2D, which encodes retinal guanylate cyclase. Ophthalmology 2000, 107, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Boon, C.J.; den Hollander, A.I.; Hoyng, C.B.; Cremers, F.P.; Klevering, B.J.; Keunen, J.E. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog. Retin. Eye Res. 2008, 27, 213–235. [Google Scholar] [CrossRef] [PubMed]
- Ba-Abbad, R.; Robson, A.G.; Yap, Y.C.; Moore, A.T.; Webster, A.R.; Holder, G.E. Prph2 mutations as a cause of electronegative ERG. Retina 2014, 34, 1235–1243. [Google Scholar] [CrossRef]
- Yamamoto, H.; Simon, A.; Eriksson, U.; Harris, E.; Berson, E.L.; Dryja, T.P. Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and fundus albipunctatus. Nat. Genet. 1999, 22, 188–191. [Google Scholar] [CrossRef]
- Schmitz-Valckenberg, S.; Pfau, M.; Fleckenstein, M.; Staurenghi, G.; Sparrow, J.R.; Bindewald-Wittich, A.; Spaide, R.F.; Wolf, S.; Sadda, S.R.; Holz, F.G. Fundus autofluorescence imaging. Prog. Retin. Eye Res. 2021, 81, 100893. [Google Scholar] [CrossRef]
- Sergouniotis, P.I.; Sohn, E.H.; Li, Z.; McBain, V.A.; Wright, G.A.; Moore, A.T.; Robson, A.G.; Holder, G.E.; Webster, A.R. Phenotypic variability in RDH5 retinopathy (Fundus Albipunctatus). Ophthalmology 2011, 118, 1661–1670. [Google Scholar] [CrossRef]
- Wang, N.K.; Chuang, L.H.; Lai, C.C.; Chou, C.L.; Chu, H.Y.; Yeung, L.; Chen, Y.P.; Chen, K.J.; Wu, W.C.; Chen, T.L.; et al. Multimodal fundus imaging in fundus albipunctatus with RDH5 mutation: A newly identified compound heterozygous mutation and review of the literature. Doc. Ophthalmol. 2012, 125, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Skalet, J.; Miyake, Y. RDH5 gene mutations and electroretinogram in fundus albipunctatus with or without macular dystrophy: RDH5 mutations and ERG in fundus albipunctatus. Doc. Ophthalmol. 2003, 107, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Krill, A.E.; Folk, M.R. Retinitis punctata albescens. A functional evaluation of an unusual case. Am. J. Ophthalmol. 1962, 53, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.A.; Roberts, M.F.; Derlacki, D.J.; Grimsby, J.L.; Yamamoto, H.; Sharon, D.; Nishiguchi, K.M.; Dryja, T.P. Novel mutations in the cellular retinaldehyde-binding protein gene (RLBP1) associated with retinitis punctata albescens: Evidence of interfamilial genetic heterogeneity and fundus changes in heterozygotes. Arch. Ophthalmol. 2004, 122, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Rattner, A.; Nathans, J. The rod photoreceptor-specific nuclear receptor Nr2e3 represses transcription of multiple cone-specific genes. J. Neurosci. 2005, 25, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Michaelides, M.; Robson, A.G.; Hawlina, M.; Vaclavik, V.; Sandbach, J.M.; Neveu, M.M.; Hogg, C.R.; Hunt, D.M.; Moore, A.T.; et al. Phenotypic variation in enhanced S-cone syndrome. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2082–2093. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Sandberg, M.A.; Caruso, R.C.; Berson, E.L.; Dryja, T.P. Shared mutations in NR2E3 in enhanced S-cone syndrome, Goldmann-Favre syndrome, and many cases of clumped pigmentary retinal degeneration. Arch. Ophthalmol. 2003, 121, 1316–1323. [Google Scholar] [CrossRef]
- Khan, A.O.; Aldahmesh, M.; Meyer, B. The enhanced S-cone syndrome in children. BMJ Case Rep. 2009, 2009, bcr1020081163. [Google Scholar] [CrossRef]
- Hull, S.; Arno, G.; Sergouniotis, P.I.; Tiffin, P.; Borman, A.D.; Chandra, A.; Robson, A.G.; Holder, G.E.; Webster, A.R.; Moore, A.T. Clinical and molecular characterization of enhanced S-cone syndrome in children. JAMA Ophthalmol. 2014, 132, 1341–1349. [Google Scholar] [CrossRef]
- Zerbib, J.; Blanco Garavito, R.; Gerber, S.; Oubraham, H.; Sikorav, A.; Audo, I.; Kaplan, J.; Rozet, J.M.; Souied, E.H. Retinochoroidal anastomosis associated with enhanced s-cone syndrome. Retin. Cases Brief Rep. 2019, 13, 295–299. [Google Scholar] [CrossRef]
- Yzer, S.; Barbazetto, I.; Allikmets, R.; van Schooneveld, M.J.; Bergen, A.; Tsang, S.H.; Jacobson, S.G.; Yannuzzi, L.A. Expanded clinical spectrum of enhanced S-cone syndrome. JAMA Ophthalmol. 2013, 131, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.K.; Fine, H.F.; Chang, S.; Chou, C.L.; Cella, W.; Tosi, J.; Lin, C.S.; Nagasaki, T.; Tsang, S.H. Cellular origin of fundus autofluorescence in patients and mice with a defective NR2E3 gene. Br. J. Ophthalmol. 2009, 93, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.K.; Lai, C.C.; Liu, C.H.; Yeh, L.K.; Chou, C.L.; Kong, J.; Nagasaki, T.; Tsang, S.H.; Chien, C.L. Origin of fundus hyperautofluorescent spots and their role in retinal degeneration in a mouse model of Goldmann-Favre syndrome. Dis. Model Mech. 2013, 6, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, S.; Li, P.; Yao, K. Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. Int. J. Mol. Sci. 2022, 23, 4883. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, V.K.L.; Takiuti, J.T.; Jauregui, R.; Mahajan, V.B.; Tsang, S.H. Rates of Bone Spicule Pigment Appearance in Patients With Retinitis Pigmentosa Sine Pigmento. Am. J. Ophthalmol. 2018, 195, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Cowing, J.A.; Michaelides, M.; Wilkie, S.E.; Jeffery, G.; Jenkins, S.A.; Mester, V.; Bird, A.C.; Robson, A.G.; Holder, G.E.; et al. Mutations in the gene KCNV2 encoding a voltage-gated potassium channel subunit cause “cone dystrophy with supernormal rod electroretinogram” in humans. Am. J. Hum. Genet. 2006, 79, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Lenis, T.L.; Dhrami-Gavazi, E.; Lee, W.; Mukkamala, S.K.; Tabacaru, M.R.; Yannuzzi, L.; Gouras, P.; Tsang, S.H. Novel Compound Heterozygous Mutations Resulting in Cone Dystrophy With Supernormal Rod Response. JAMA Ophthalmol. 2013, 131, 1482–1485. [Google Scholar] [CrossRef]
- Abdelkader, E.; Yasir, Z.H.; Khan, A.M.; Raddadi, O.; Khandekar, R.; Alateeq, N.; Nowilaty, S.; AlShahrani, N.; Schatz, P. Analysis of retinal structure and function in cone dystrophy with supernormal rod response. Doc. Ophthalmol. 2020, 141, 23–32. [Google Scholar] [CrossRef]
- Liu, P.K.; Ryu, J.; Yeh, L.K.; Chen, K.J.; Tsang, S.H.; Liu, L.; Wang, N.K. A novel KCNV2 mutation in a patient taking hydroxychloroquine associated with cone dystrophy with supernormal rod response. Ophthalmic Genet. 2021, 42, 458–463. [Google Scholar] [CrossRef]
- Ding, H.J.; Denniston, A.K.; Rao, V.K.; Gordon, C. Hydroxychloroquine-related retinal toxicity. Rheumatology 2015, 55, 957–967. [Google Scholar] [CrossRef]
- Chen, E.; Brown, D.M.; Benz, M.S.; Fish, R.H.; Wong, T.P.; Kim, R.Y.; Major, J.C. Spectral domain optical coherence tomography as an effective screening test for hydroxychloroquine retinopathy (the “flying saucer” sign). Clin. Ophthalmol. 2010, 4, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.T.; Costenbader, K.H.; Desmarais, J.; Ginzler, E.M.; Fett, N.; Goodman, S.M.; O’Dell, J.R.; Schmajuk, G.; Werth, V.P.; Melles, R.B.; et al. American College of Rheumatology, American Academy of Dermatology, Rheumatologic Dermatology Society, and American Academy of Ophthalmology 2020 Joint Statement on Hydroxychloroquine Use With Respect to Retinal Toxicity. Arthritis Rheumatol. 2021, 73, 908–911. [Google Scholar] [CrossRef] [PubMed]
- Koh, V.; Tan, C.; Nah, G.; Zhao, P.; Yang, A.; Lin, S.T.; Wong, T.Y.; Saw, S.M.; Chia, A. Correlation of structural and electrophysiological changes in the retina of young high myopes. Ophthalmic Physiol. Opt. 2014, 34, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Sachidanandam, R.; Ravi, P.; Sen, P. Effect of axial length on full-field and multifocal electroretinograms. Clin. Exp. Optom. 2017, 100, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Chakraborty, R.; Verkicharla, P.K. Correction to: Electroretinogram responses in myopia: A review. Doc. Ophthalmol. 2022, 145, 97–98. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Xu, Z.; Mei, M.; Chen, Z.; Wang, K.; Liu, Y.; Tang, T.; Priydarshi, M.K.; Meng, X.; Zhao, S.; et al. Soft transparent graphene contact lens electrodes for conformal full-cornea recording of electroretinogram. Nat. Commun. 2018, 9, 2334. [Google Scholar] [CrossRef] [PubMed]
- Dockery, A.; Whelan, L.; Humphries, P.; Farrar, G.J. Next-Generation Sequencing Applications for Inherited Retinal Diseases. Int. J. Mol. Sci. 2021, 22, 5684. [Google Scholar] [CrossRef]
- Fadaie, Z.; Khan, M.; Del Pozo-Valero, M.; Cornelis, S.S.; Ayuso, C.; Cremers, F.P.M.; Roosing, S.; The Abca Study, G. Identification of splice defects due to noncanonical splice site or deep-intronic variants in ABCA4. Hum. Mutat. 2019, 40, 2365–2376. [Google Scholar] [CrossRef]
- Qian, X.; Wang, J.; Wang, M.; Igelman, A.D.; Jones, K.D.; Li, Y.; Wang, K.; Goetz, K.E.; Birch, D.G.; Yang, P.; et al. Identification of Deep-Intronic Splice Mutations in a Large Cohort of Patients With Inherited Retinal Diseases. Front. Genet. 2021, 12, 647400. [Google Scholar] [CrossRef]
- Cornish, E.E.; Vaze, A.; Jamieson, R.V.; Grigg, J.R. The electroretinogram in the genomics era: Outer retinal disorders. Eye 2021, 35, 2406–2418. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.; Lange, C.; Laird, N.M.; Won, S.; Hersh, C.P.; Morrow, J.; Hobbs, B.D.; Lutz, S.M.; Ruczinski, I.; Beaty, T.H.; et al. Gene-based segregation method for identifying rare variants in family-based sequencing studies. Genet. Epidemiol. 2017, 41, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Jayanna, S.; Jalali, S.; Padhi, T.R.; Agarwal, K.; Chhablani, J. OCT Imaging in Infants. Semin Ophthalmol. 2022, 37, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Magnusdottir, V.; Vehmeijer, W.B.; Eliasdottir, T.S.; Hardarson, S.H.; Schalij-Delfos, N.E.; Stefánsson, E. Fundus imaging in newborn children with wide-field scanning laser ophthalmoscope. Acta Ophthalmol. 2017, 95, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, F.; Parkinson, J.E.; Lalonde, M.R. Anesthesia and Electroretinography. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1897. [Google Scholar]
- Mahroo, O.A. Visual electrophysiology and “the potential of the potentials”. Eye 2023, 37, 2399–2408. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.; Gordon-Reid, A.; Shawkat, F.; Self, J.E. Comparison of the handheld RETeval ERG system with a routine ERG system in healthy adults and in paediatric patients. Eye 2021, 35, 2180–2189. [Google Scholar] [CrossRef] [PubMed]
- You, J.Y.; Dorfman, A.L.; Gauvin, M.; Vatcher, D.; Polomeno, R.C.; Little, J.M.; Lachapelle, P. Comparing the RETeval® portable ERG device with more traditional tabletop ERG systems in normal subjects and selected retinopathies. Doc. Ophthalmol. 2023, 146, 137–150. [Google Scholar] [CrossRef]
- Glinton, S.L.; Calcagni, A.; Lilaonitkul, W.; Pontikos, N.; Vermeirsch, S.; Zhang, G.; Arno, G.; Wagner, S.K.; Michaelides, M.; Keane, P.A.; et al. Phenotyping of ABCA4 Retinopathy by Machine Learning Analysis of Full-Field Electroretinography. Transl. Vis. Sci. Technol. 2022, 11, 34. [Google Scholar] [CrossRef]
- Gajendran, M.K.; Rohowetz, L.J.; Koulen, P.; Mehdizadeh, A. Novel Machine-Learning Based Framework Using Electroretinography Data for the Detection of Early-Stage Glaucoma. Front. Neurosci. 2022, 16, 869137. [Google Scholar] [CrossRef]
- Schwitzer, T.; Leboyer, M.; Laprévote, V.; Louis Dorr, V.; Schwan, R. Using retinal electrophysiology toward precision psychiatry. Eur. Psychiatry 2022, 65, e9. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
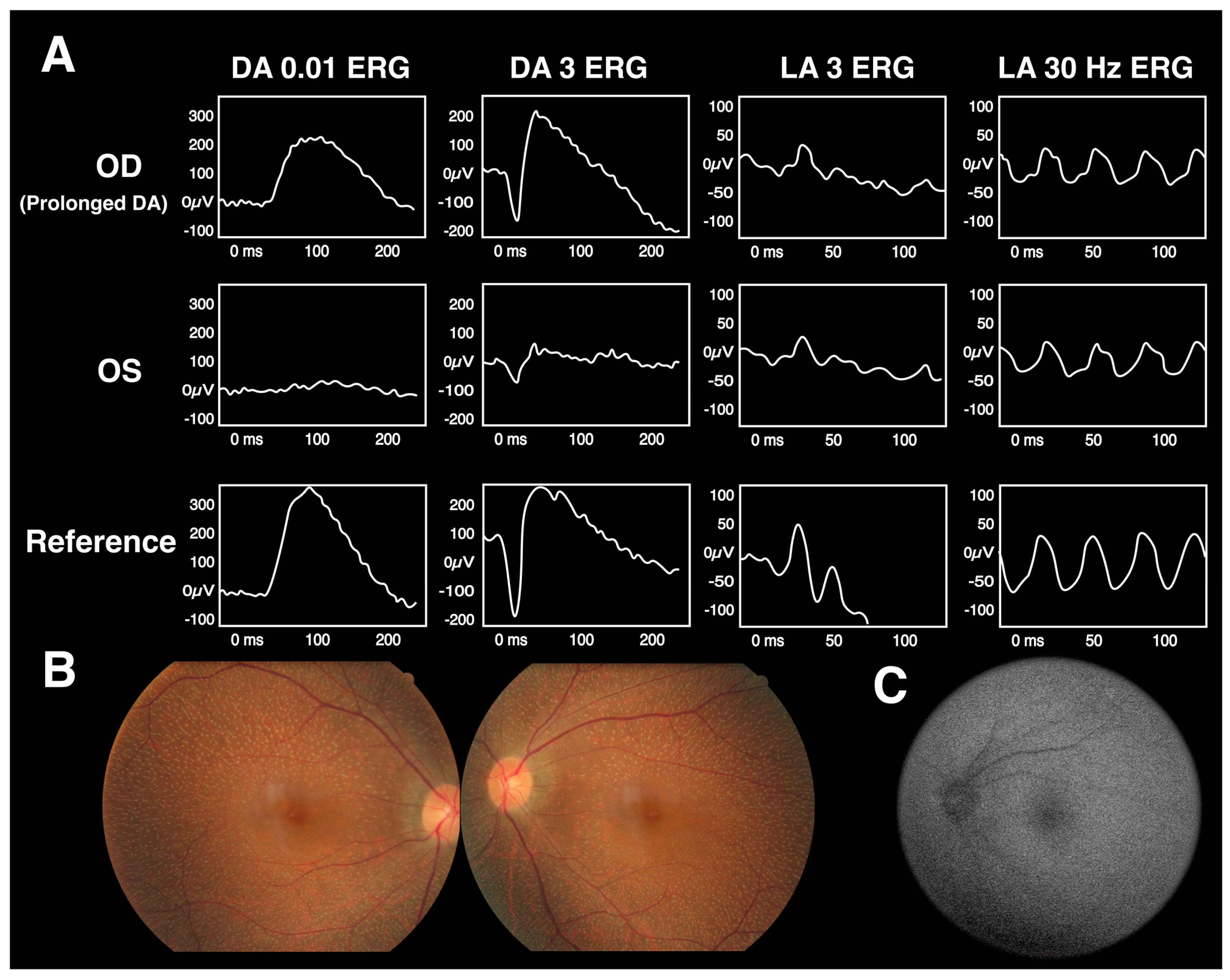{kind=link}
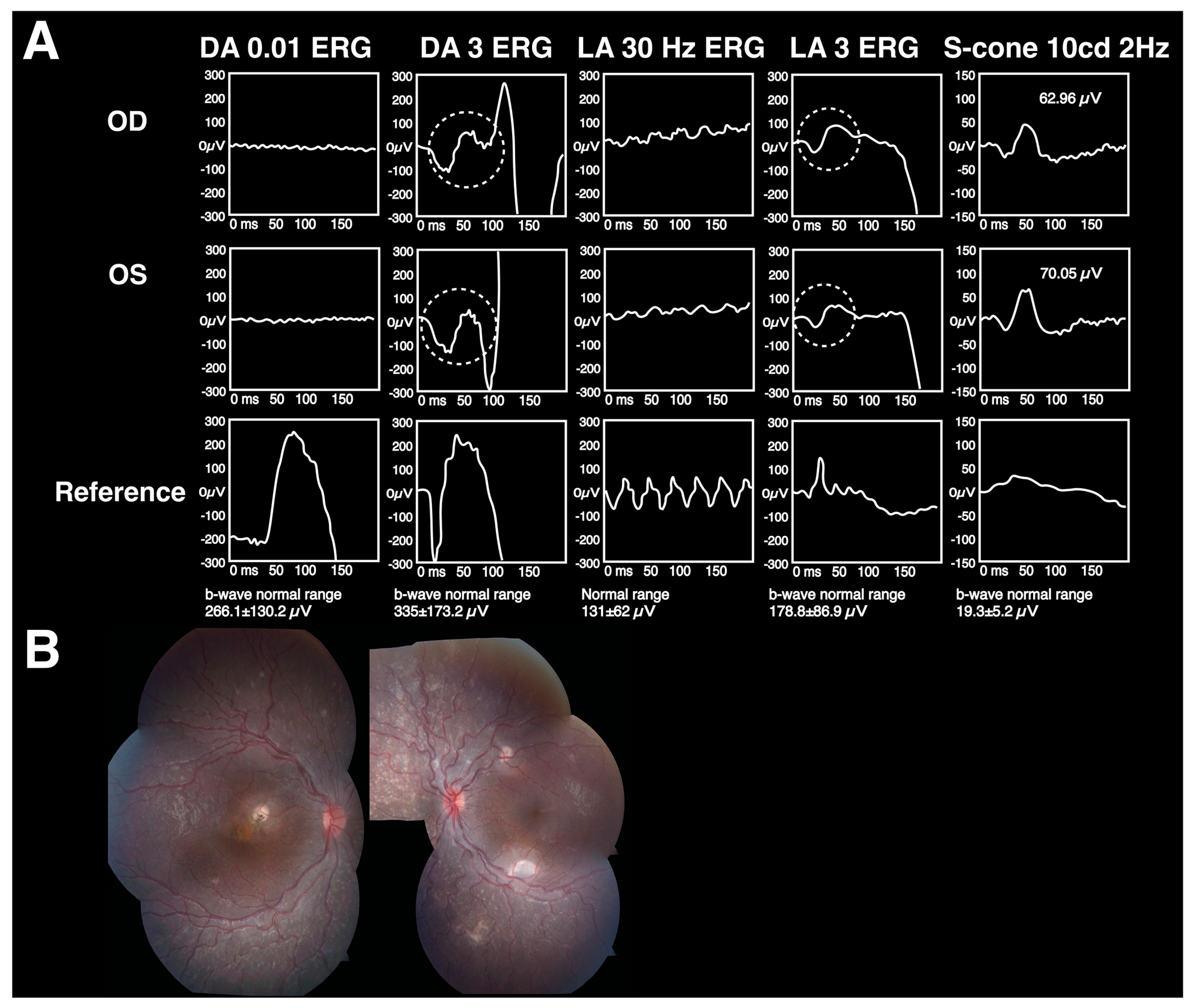{kind=link}
{kind=link}
| Congenital Stationary Night Blindness (Mutated Genes) | |||||
|---|---|---|---|---|---|
| Dysfunctional retinal cells | Bipolar cells | iCSBN (CACNA1F) cCSNB (NYX) | N/A | iCSBN (CABP4, CACNA2D4) cCSNB (GPR170, GRM6, LRIT3, TRPM1) | N/A |
| Rods | N/A | Riggs (GNAT1, PDE6B, RHO) | Riggs (SLC24A1) | Oguchi (SAG, GRK1) | |
| RPE | N/A | N/A | N/A | Fundus albipunctatus (RDH5) | |
| Fundus appearance | Normal fundus | Abnormal fundus | |||
| Inheritance pattern | X-linked recessive | Autosomal dominant | Autosomal recessive | ||
| Disease | Ocular/Fundus Abnormalities | Inheritance Patterns: Mutated Genes | Scotopic ERG | Photopic ERG | Comments | ||
|---|---|---|---|---|---|---|---|
| DA 0.01 ERG | DA 3 or 10 ERG | LA 3 ERG | LA 30 Hz | ||||
| |||||||
| Incomplete Congenital Stationary Night Blindness (iCSNB) |
|
| ↓ (“Incompletely” abolished) | ↓ b-wave (-Ve, but not required) | ↓b-wave |
|
|
| Complete Congenital Stationary Night Blindness (cCSNB) |
|
| ↓↓ Extinguished (“Completely” abolished) | ↓ b-wave (-Ve, but not required) |
| Normal, possible ↑ implicit time |
|
| Riggs-type CSNB |
|
| ↓↓ Extinguished | ↓ a-wave & ↓ b-wave (Possibly -Ve) | Normal |
| |
| X-linked Retinoschisis (XLRS) |
| XR: RS1 | ↓ | ↓ b-wave (-Ve, but not required) | ↓ | ↓ |
|
| Cone–rod Dystrophy |
| AD: CRX, GUCY2D, PRPH2, RAX2 | ↓ | ↓ b-wave (-Ve) | ↓↓ |
| |
| |||||||
| Fundus Albipunctatus |
| AR: RDH5 |
| Varies |
| ||
| Enhanced S-cone Syndrome (ESCS)/ Goldmann-Favre Syndrome |
| AR: NR2E3 | ↓↓Extinguished (∵ cone-dominated retina) |
|
| ||
| Cone Dystrophy with Supernormal Rod Response (CDSRR) |
| AR: KCNV2 | ↓ and delayed | ↑ b-wave disproportionally | ↓↓ (Cones more affected than rods) |
| |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, T.-H.; Kang, E.Y.-C.; Lin, P.-H.; Wu, P.-L.; Sachs, J.A.; Wang, N.-K. The Value of Electroretinography in Identifying Candidate Genes for Inherited Retinal Dystrophies: A Diagnostic Guide. Diagnostics 2023, 13, 3041. https://doi.org/10.3390/diagnostics13193041
Yang T-H, Kang EY-C, Lin P-H, Wu P-L, Sachs JA, Wang N-K. The Value of Electroretinography in Identifying Candidate Genes for Inherited Retinal Dystrophies: A Diagnostic Guide. Diagnostics. 2023; 13(19):3041. https://doi.org/10.3390/diagnostics13193041
Chicago/Turabian StyleYang, Tsai-Hsuan, Eugene Yu-Chuan Kang, Pei-Hsuan Lin, Pei-Liang Wu, Jacob Aaron Sachs, and Nan-Kai Wang. 2023. "The Value of Electroretinography in Identifying Candidate Genes for Inherited Retinal Dystrophies: A Diagnostic Guide" Diagnostics 13, no. 19: 3041. https://doi.org/10.3390/diagnostics13193041
APA StyleYang, T. -H., Kang, E. Y. -C., Lin, P. -H., Wu, P. -L., Sachs, J. A., & Wang, N. -K. (2023). The Value of Electroretinography in Identifying Candidate Genes for Inherited Retinal Dystrophies: A Diagnostic Guide. Diagnostics, 13(19), 3041. https://doi.org/10.3390/diagnostics13193041