
Pitfalls of Thrombotic Microangiopathies in Children: Two Case Reports and Literature Review
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Case Reports
2.2. Literature Review and Data Extraction
3. Results
3.1. Case 1—aHUS
3.2. Case 2—TTP
3.3. Literature Research
4. Discussion
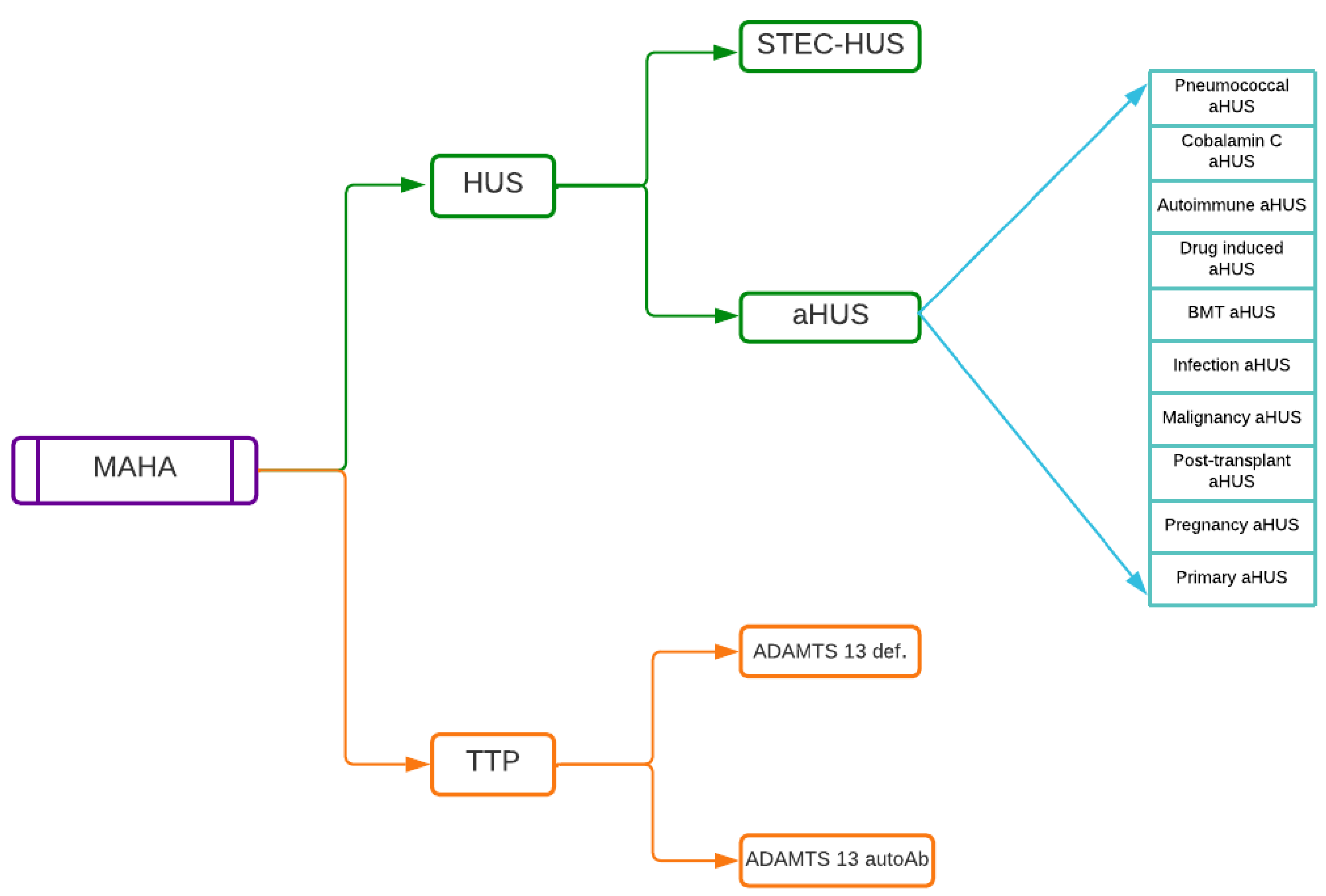
4.1. aHUS
4.2. Clinical and Molecular Diagnosis
4.3. TTP
4.4. COVID-19 and Thrombotic Microangiopathies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chiasakul, T.; Cuker, A. Clinical and laboratory diagnosis of TTP: An integrated approach. Hematology 2018, 2018, 530–538. [Google Scholar] [CrossRef] [Green Version]
- Joly, B.S.; Coppo, P.; Veyradier, A. Thrombotic thrombocytopenic purpura. Blood 2017, 129, 2836–2846. [Google Scholar] [CrossRef] [Green Version]
- Sayani, F.A.; Abrams, C.S. How I treat refractory thrombotic thrombocytopenic purpura. Blood 2015, 125, 3860–3867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neunert, C.; Lim, W.; Crowther, M.; Cohen, A.; Solberg, L. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011, 117, 4190–4207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufeld, J.; Reinhardt, M.; Schroder, C.; Brasen, J.H.; Wiech, T.; Brylka, P.; Khaled, A.; Bergmann, C.; Haller, H.; Gackler, A.; et al. Atypical HUS triggered by infection with SARS-CoV2. Kidney Int. Rep. 2021, 6, 2709–2712. [Google Scholar] [CrossRef] [PubMed]
- Mat, O.; Ghisdal, L.; Massart, A.; Aydin, S.; Goubella, A.; Blankoff, N.; Gankam, F.; Debelle, F.; Mat, Q. Kidney thrombotic microangiopathy after COVID-19 associated with C3 gene mutation. Kidney Int. Rep. 2021, 6, 1732–1737. [Google Scholar]
- Ville, S.; Le Bot, S.; Chapelet-Debout, A.; Blancho, G.; Fremeaux-Bacchi, V.; Deltombe, C.; Fakhouri, F. Atypical HUS relapse triggered by COVID-19. Kidney Int. 2021, 99, 267–268. [Google Scholar] [CrossRef]
- Diorio, C.; McNerney, K.O.; Lambert, M.; Paessler, M.; Anderson, E.M.; Henrickson, S.E.; Chase, J.; Liebling, E.J.; Burudpakdee, C.; Lee, J.H.; et al. Evidence of thrombotic microangiopathy in children with SARS-CoV-2 across the spectrum of clinical presentations. Blood Adv. 2020, 4, 6051–6063. [Google Scholar] [CrossRef]
- Galeotti, C.; Bayry, J. Autoimmune and inflammatory diseases following COVID-19. Nat. Rev. Rheumatol. 2020, 16, 413–414. [Google Scholar] [CrossRef]
- Bendapudi, P.K.; Hurwitz, S.; Fry, A.; Marques, M.B.; Waldo, S.W.; Li, A.; Sun, L.; Upadhyay, V.; Hamdan, A.; Brunner, A.M.; et al. Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: A cohort study. Lancet Haematol. 2017, 4, e157–e164. [Google Scholar] [CrossRef]
- Miller, D.D.; Krenzer, J.A.; Kenkre, V.P.; Rose, W.N. Sequential immune thrombocytopenia (ITP) and thrombotic thrombocytopenic purpura (TTP) in an elderly male patient with primary Sjogren’s syndrome: When in doubt, use the PLASMIC Score. Case Rep. Med. 2021, 2021, 6869342. [Google Scholar] [CrossRef] [PubMed]
- Kasturiarachi, B.M.; Alsbrook, D.L.; Crook, J.; Shah, N. An Immunologic Storm: A Case of Encephalitis and Thrombotic Thrombocytopenic Purpura With Underlying Likely Sjogren’s Syndrome Induced by a COVID-19 Immune Response. Neurohospitalist 2022, 12, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Vorster, L.; Kirk, S.E.; Muscal, E.; Despotovic, J.M.; Cohen, C.T.; Sartain, S.E. COVID-19 vaccine (mRNA BNT162b2) and COVID-19 infection-induced thrombotic thrombocytopenic purpura in adolescents. Pediatr. Blood Cancer 2022, 69, e29681. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Rojas, J.; Campano, W.; Tasayco, J.; Siu-Lam, A.; Ortega-Ocas, C.; Atamari-Anahui, N. Thrombotic thrombocytopenic purpura associated with COVID-19 in a critically ill child: A Peruvian case report. Bol. Med. Hosp. Infant. Mex. 2022, 79, 123–128. [Google Scholar] [CrossRef]
- Kirpalani, A.; Garabon, J.; Amos, K.; Patel, S.; Sharma, A.P.; Ganesan, S.L.; Barton, M.; Cacciotti, C.; Leppington, S.; Bakovic, L.; et al. Thrombotic thrombocytopenic purpura temporally associated with BNT162b2 vaccination in an adolescent successfully treated with caplacizumab. Br. J. Haematol. 2022, 196, e11–e14. [Google Scholar] [CrossRef]
- Hamza, S.B. Severe SARS-COV-2 infection in pediatric patient with atypical Hemolytic Uremic Syndrome: A case report. Ann. Med. Surg. 2022, 75, 103400. [Google Scholar] [CrossRef]
- Khandelwal, P.; Krishnasamy, S.; Govindarajan, S.; Kumar, M.; Marik, B.; Sinha, A.; Hari, P.; Bagga, A. Anti-factor H antibody associated hemolytic uremic syndrome following SARS-CoV-2 infection. Pediatr. Nephrol. 2022, 37, 2151–2156. [Google Scholar] [CrossRef]
- Dalkıran, T.; Kandur, Y.; Kara, E.M.; Dağoğlu, B.; Taner, S.; Öncü, D. Thrombotic Microangiopathy in a Severe Pediatric Case of COVID-19. Clinical medicine insights. Pediatrics 2021, 15, 11795565211049897. [Google Scholar]
- Searcy, K.; Jagadish, A.; Pichilingue-Reto, P.; Baliga, R. Coronavirus Disease 2019 (COVID-19) Associated Hemolytic Uremic Syndrome in a Toddler. Case Rep. Pediatr. 2022, 2022, 3811170. [Google Scholar] [CrossRef]
- Alizadeh, F.; O’Halloran, A.; Alghamdi, A.; Chen, C.; Trissal, M.; Traum, A.; DeCourcey, D. Toddler with New Onset Diabetes and Atypical Hemolytic-Uremic Syndrome in the Setting of COVID-19. Pediatrics 2021, 147, e2020016774. [Google Scholar] [CrossRef]
- Goodship, T.H.J.; Cook, H.T.; Fakhouri, F.; Fervenza, F.C.; Frémeaux-Bacchi, V.; Kavanagh, D.; Zipfel, P.F. Atypical hemolytic uremic syndrome and C3 glomerulopathy: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017, 91, 539–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loirat, C.; Fakhouri, F.; Ariceta, G.; Besbas, N.; Bitzan, M.; Bjerre, A.; Coppo, R.; Emma, F.; Johnson, S.; Karpman, D.; et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr. Nephrol. 2016, 31, 15–39. [Google Scholar] [CrossRef] [PubMed]
- Fremeaux-Bacchi, V.; Fakhouri, F.; Garnier, A.; Bienaimé, F.; Dragon-Durey, M.A.; Ngo, S.; Moulin, B.; Servais, A.; Provot, F.; Rostaing, L.; et al. Genetics and outcome of atypical hemolytic uremic syndrome: A nationwide French series comparing children and adults. Clin. J. Am. Soc. Nephrol. 2013, 8, 554–562. [Google Scholar] [CrossRef] [Green Version]
- Maga, T.K.; Nishimura, C.J.; Weaver, A.E.; Frees, K.L.; Smith, R.J. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum. Mutat. 2010, 31, E1445–E1460. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, D.; Goodship, T.H.; Richards, A. Atypical hemolytic uremic syndrome. Semin. Nephrol. 2013, 33, 508–530. [Google Scholar] [CrossRef] [Green Version]
- Thurman, J.M. Complement and the Kidney: An Overview. Adv. Chronic. Kidney Dis. 2020, 27, 86–94. [Google Scholar] [CrossRef]
- Noris, M.; Caprioli, J.; Bresin, E.; Mossali, C.; Pianetti, G.; Gamba, S.; Daina, E.; Fenili, C.; Castelletti, F.; Sorosina, A.; et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin. J. Am. Soc. Nephrol. 2010, 5, 1844–1859. [Google Scholar] [CrossRef] [Green Version]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar] [CrossRef] [Green Version]
- Rother, R.P.; Rollins, S.A.; Mojcik, C.F.; Brodsky, R.A.; Bell, L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat. Biotechnol. 2007, 25, 1256–1264. [Google Scholar] [CrossRef]
- Scully, M.; Cataland, S.; Coppo, P.; de la Rubia, J.; Friedman, K.D.; Hovinga, J.K.; Lämmle, B.; Matsumoto, M.; Pavenski, K.; Sadler, E.; et al. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J. Thromb. Haemost. 2017, 15, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.L.; Vesely, S.K.; Cataland, S.R.; Coppo, P.; Geldziler, B.; Iorio, A.; Matsumoto, M.; Mustafa, R.A.; Pai, M.; Rock, G.; et al. ISTH guidelines for treatment of thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2020, 18, 2496–2502. [Google Scholar] [CrossRef] [PubMed]
- Bell, W.R.; Braine, H.G.; Ness, P.M.; Kickler, T.S. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: Clinical experience in 108 patients. N. Engl. J. Med. 1991, 325, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Cataland, S.R.; Kourlas, P.J.; Yang, S.; Geyer, S.; Witkoff, L.; Wu, H.; Masias, C.; George, J.N.; Wu, H.M. Cyclosporine or steroids as an adjunct to plasma exchange in the treatment of immune-mediated thrombotic thrombocytopenic purpura. Blood Adv. 2017, 1, 2075–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucukyurt, S.; Eskazan, A.E. Assessment and monitoring of patients with immune-mediated thrombotic thrombocytopenic purpura (iTTP): Strategies to improve outcomes. J. Blood Med. 2020, 11, 319–326. [Google Scholar] [CrossRef]
- Ulrichts, H.; Silence, K.; Schoolmeester, A.; de Jaegere, P.; Rossenu, S.; Roodt, J.; Priem, S.; Lauwereys, M.; Casteels, P.; Van Bockstaele, F.; et al. Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared with currently marketed antiplatelet drugs. Blood 2011, 118, 757–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyvandi, F.; Scully, M.; Kremer Hovinga, J.A.; Cataland, S.; Knöbl, P.; Wu, H.; Artoni, A.; Westwood, J.-P.; Mansouri Taleghani, M.; Jilma, B.; et al. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2016, 374, 511–522. [Google Scholar] [CrossRef]
- Scully, M.; Goodship, T. How I treat thrombotic thrombocytopenic purpura and atypical haemolytic uraemic syndrome. Br. J. Haematol. 2014, 164, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Lazaruc, T.I.; Bodescu Amancei Ionescu, L.; Lupu, V.V.; Muntean, C.; Bogos, R.A.; Ivanov, A.; Scurtu, G.; Starcea, I.M.; Miron, I.C.; Mocanu, M.A. Thrombosis in Chronic Kidney Disease in Children. Diagnostics 2022, 12, 2931. [Google Scholar] [CrossRef]
- Pouletty, M.; Borocco, C.; Ouldali, N.; Caseris, M.; Basmaci, R.; Lachaume, N.; Bensaid, P.; Pichard, S.; Kouider, H.; Morelle, G.; et al. Paediatric multisystem inflammatory syndrome temporally associated with SARS-CoV-2 mimicking kawasaki disease (kawa-COVID-19): A multicentre cohort. Ann. Rheum. Dis. 2020, 79, 999–1006. [Google Scholar] [CrossRef]
- Esposito, S.; Principi, N. Multisystem inflammatory syndrome in children related to SARS-CoV-2. Pediatr. Drugs 2021, 23, 119–129. [Google Scholar] [CrossRef]
- Richardson, G.M.; Su, S.W.; Iragorri, S. Case report: Diarrhea-associated hemolytic uremic syndrome in the Era of COVID-19. Front. Pediatr. 2022, 10, 979850. [Google Scholar] [CrossRef] [PubMed]
- Van Quekelberghe, C.; Latta, K.; Kunzmann, S.; Grohmann, M.; Hansen, M. Atypical hemolytic uremic syndrome induced by SARS-CoV2 infection in infants with EXOSC3 mutation. Pediatr. Nephrol. 2022, 37, 2781–2784. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Yu, J.; Yuan, X.; Chen, H.; Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood 2020, 136, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.M.; Kahwash, R. Will complement inhibition be the new target in treating COVID-19-related systemic thrombosis? Circulation. 2020, 141, 1739–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrill, J.T.; Erkan, D.; Winakur, J.; James, J.A. Emerging evidence of a COVID-19 thrombotic syndrome has treatment implications. Nat. Rev. Rheumatol. 2020, 16, 581–589. [Google Scholar] [CrossRef]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef]
- Gralinski, L.E.; Sheahan, T.P.; Morrison, T.E.; Menachery, V.D.; Je Sen, K.; Leist, S.R.; Whitmore, A.; Heise, M.T.; Baric, R.S. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio 2018, 9, e01753-18. [Google Scholar] [CrossRef] [Green Version]
- Pavord, S.; Scully, M.; Hunt, B.J.; Lester, W.; Bagot, C.; Craven, B.; Rampotas, A.; Ambler, G.; Makris, M. Clinical features of vaccine-induced immune thrombocytopenia and thrombosis. N. Engl. J. Med. 2021, 385, 1680–1689. [Google Scholar] [CrossRef]
- McGonagle, D.; De Marco, G.; Bridgewood, C. Mechanisms of immunothrombosis in vaccine-induced thrombotic thrombocytopenia (VITT) compared to natural SARS-CoV-2 infection. J. Autoimmun. 2021, 121, 102662. [Google Scholar]
- Szóstek-Mioduchowska, A.; Kordowitzki, P. Shedding light on the possible link between ADAMTS13 and vaccine—Induced thrombotic thrombocytopenia. Cells 2021, 10, 2785. [Google Scholar] [CrossRef] [PubMed]
- Petri, A.; Kim, H.J.; Xu, Y.; de Groot, R.; Li, C.; Vandenbulcke, A.; Vanhoorelbeke, K.; Emsley, J.; Crawley, J.T.B. Crystal structure and substrate-induced activation of ADAMTS13. Nat. Commun. 2019, 10, 3781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matošević, M.; Kos, I.; Davidović, M.; Ban, M.; Matković, H.; Jakopčić, I.; Brinar, I.V.; Szilágyi, A.; Csuka, D.; Sinkovits, G.; et al. Hemolytic uremic syndrome in the setting of COVID-19 successfully treated with complement inhibition therapy: An instructive case report of a previously healthy toddler and review of literature. Front. Pediatr. 2023, 11, 154. [Google Scholar] [CrossRef]
- Raina, R.; Vijayvargiya, N.; Khooblall, A.; Melachuri, M.; Deshpande, S.; Sharma, D.; Mathur, K.; Arora, M.; Sethi, S.K.; Sandhu, S. Pediatric atypical hemolytic uremic syndrome advances. Cells 2021, 10, 3580. [Google Scholar] [CrossRef] [PubMed]
- Diorio, C.; Shraim, R.; Vella, L.A.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Canna, S.W.; Henrickson, S.E.; McNerney, K.O.; Balamuth, F.; et al. Proteomic profiling of MIS-C patients indicates heterogeneity relating to interferon gamma dysregulation and vascular endothelial dysfunction. Nat. Commun. 2021, 12, 7222. [Google Scholar] [CrossRef]
- Shah, H.; Kim, A.; Sukumar, S.; Mazepa, M.; Kohli, R.; Braunstein, E.M.; Brodsky, R.A.; Cataland, S.; Chaturvedi, S. SARS-CoV-2 vaccination and immune thrombotic thrombocytopenic purpura. Blood 2022, 139, 2570–2573. [Google Scholar] [CrossRef]
- Picod, A.; Rebibou, J.M.; Dossier, A.; Cador, B.; Ribes, D.; Vasco-Moynet, C.; Stephan, C.; Bellal, M.; Wynckel, A.; Poullin, P.; et al. Immune-mediated thrombotic thrombocytopenic purpura following COVID-19 vaccination. Blood 2022, 139, 2565–2569. [Google Scholar] [CrossRef]
- Verma, D.P.; Dandu, H.; Yadav, G.; Verma, S.P. Complicated case of COVID-19 disease with overlapping features of thrombotic thrombocytopenic purpura and haemophagocytic lymphohistiocytosis. BMJ Case Rep. 2021, 14, e242202. [Google Scholar] [CrossRef]
- Hidalgo Filho, C.M.T.; Dalessandro Adamo, D.S.M.; Lopes, C.M.; Martin, E.M. Thrombotic thrombocytopenic purpura associated with COVID-19 in a pediatric patient: Case report. Hematol. Transfus Cell Ther. 2021, 43, 349–352. [Google Scholar] [CrossRef]
- Zhang, J.; Garrett, S.; Sun, J. Gastrointestinal symptoms, pathophysiology, and treatment in COVID-19. Genes Dis. 2021, 8, 385–400. [Google Scholar] [CrossRef]
- Malgaj Vrečko, M.; Aleš Rigler, A.; Večerić-Haler, Ž. Coronavirus Disease 2019-Associated Thrombotic Microangiopathy: Literature Review. Int. J. Mol. Sci. 2022, 23, 11307. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Liver function |
|
| Inflammatory syndrome |
|
| Blood culture |
|
| Ionogram |
|
| Complement |
|
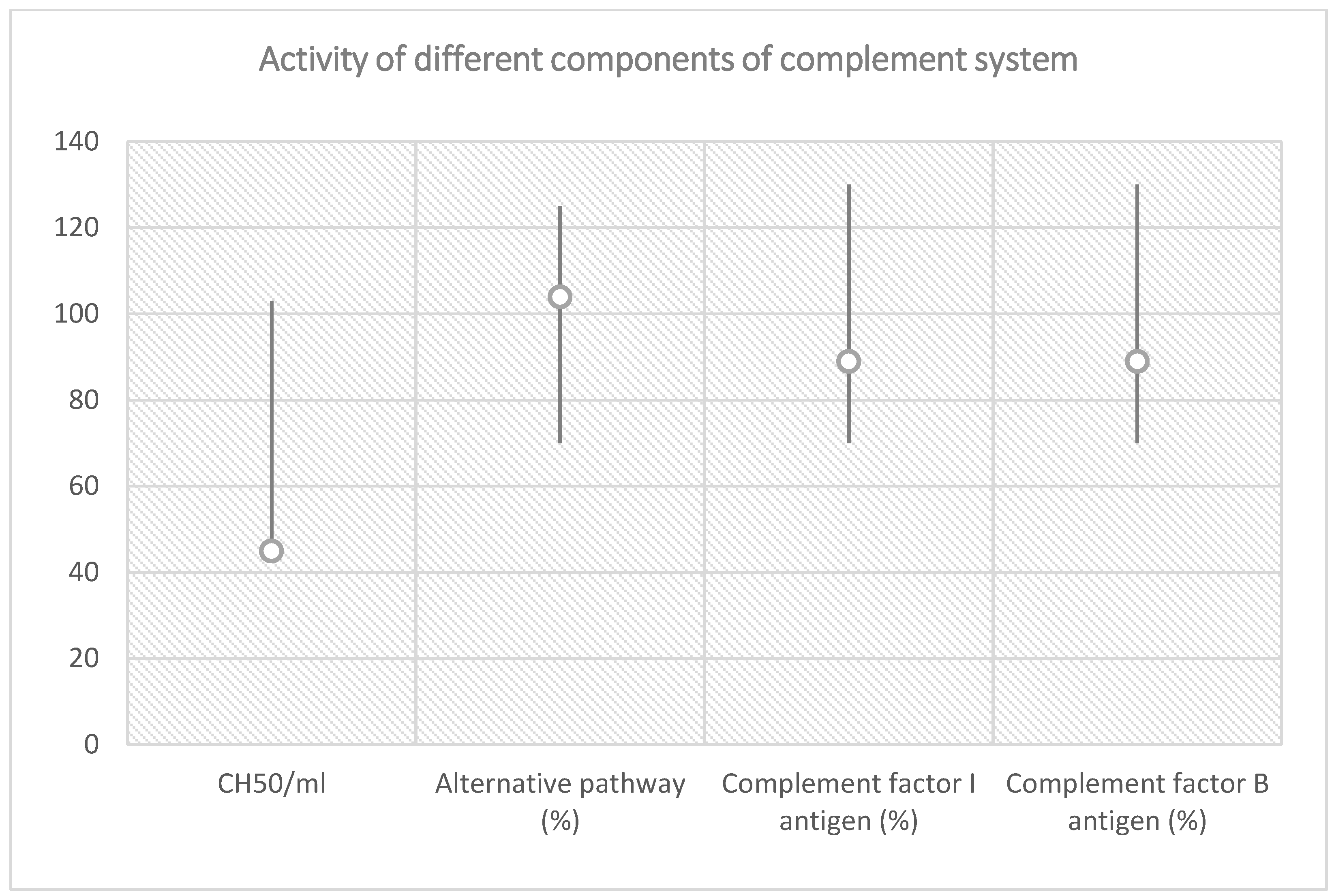
| Total Complement Activity |
|---|
| Classical pathway (hemolytic test): 45 CH50/mL (reference interval 48–103 CH50/mL) |
| Alternative pathway (WIELISA-Alt): 104% (reference interval 70–125%) |
| Complement C3: 0.71 g/L (reference interval 0.9–1.8 g/L) |
| Complement C4: 0.08 g/L (reference interval: 0.15–0.55 g/L) |
| Factor H antigen: 375 mg/L (reference interval 250–880 mg/L) |
| Complement factor I antigen: 89% (reference interval 70–130%) |
| Complement factor B antigen: 92% (reference interval 70–130%) |
| Anti-factor H IgG autoantibody: 16 AU/mL (reference interval < 110 AU/mL) |
| C1q antigen = 58 mg/L (reference interval 60–180 mg/L) |
| Anti-C1q IgG autoantibody = 0 U/mL (reference interval < 52 U/mL) |
| Haptoglobin < 0.07 g/L (reference interval 0.3–2.0 g/L) |
| Criteria and Points | |
|---|---|
| Platelet count < 30 × 109 per L | 1 |
| Hemolysis variable | 1 |
| No active evidence of cancer | 1 |
| No history of stem-cell or solid-organ transplant | 1 |
| Mean Corpuscular Volume < 90 fL | 0 |
| International Normalized Ratio < 1.5 | 1 |
| Creatinine < 2.0 mg/dL | 1 |
| Parameter | Points |
|---|---|
| Creatinine < 2.26 mg/dL | 1 |
| Platelet count < 30 × 109/L | 1 |
| Positive anti-nuclear antibodies (ANA) | 0 |
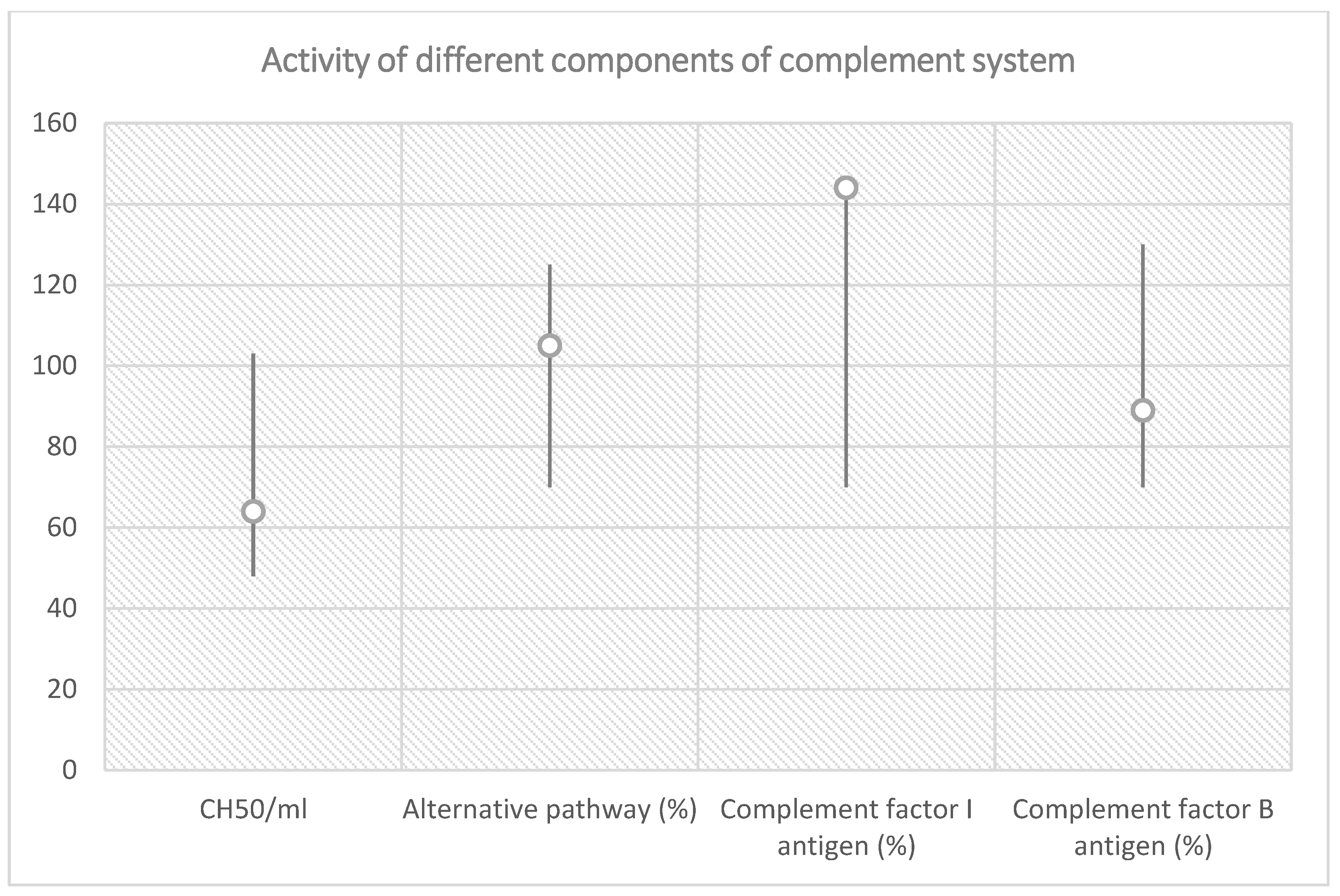
| Total Complement Activity |
|---|
| Classical pathway (hemolytic test): 64 CH50/mL (reference range 48–103 CH50/mL) |
| Alternative pathway (WIELISA-Alt): 105% (reference range 70–125%) |
| Complement C3: 1.41 g/L (reference range 0.9–1.8 g/L) |
| Complement C4: 0.33 g/L (reference range: 0.15–0.55 g/L) |
| Factor H antigen: 802 mg/L (reference range 250–880 mg/L) |
| Complement factor I antigen: 144% (reference range 70–130%) |
| Complement factor B antigen: 92% (reference range 70–130%) |
| Anti-factor H IgG autoantibody: 14 AU/mL (reference range < 110 AU/mL) |
| C1q antigen = 102 mg/L (reference range 60–180 mg/L) |
| Anti-C1q IgG autoantibody = 3 U/mL (reference range < 52 U/mL) |
| Haptoglobin < 0.07 g/L (reference range 0.3–2.0 g/L) |
| Gene | Protein Function a | Frequency in a Large Italian Cohort Study (n = 214) b | Frequency in a Large French Cohort Study (n = 144) c |
|---|---|---|---|
| FH | Cofactor for inactivation of C3b Inhibits the assembly of C3 and C5 convertases | 24% | 27% |
| FI | Cleaves and inactivates C3b | 4% | 8% |
| MCP | Cofactor for inactivation of C3b | 7% | 9% |
| C3 | Initiates alternative pathway of complement | 4% | 8% |
| FB | Links to C3, forming C3 convertase | <1% | 2 |
| THBD | Complement inactivation together with FI | 5% | n/a |
| Mutations in more than 1 gene | 3% | 4% | |
| No identified abnormality | 49% | 34% | |
| Anti-FH antibodies | 3% | 6% | |
| TTP | Characteristics | aHUS |
| ADAMTS13 deficiency | Mechanism | Defect in complement regulation |
| <10% | Serum ADAMTS13 activity | >10% |
| Maximal incidence under 40 years | Age | Mainly childhood, also possible in adults |
| Moderate/severe | Thrombocytopenia | Moderate/severe |
| Present | MAHA | Present |
| Atypical | Renal failure | Typical |
| Typical | Neurologic symptoms | Atypical |
| Possible | Fever | Possible |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mocanu, A.; Bogos, R.A.; Lazaruc, T.I.; Cianga, A.L.; Lupu, V.V.; Ioniuc, I.; Alecsa, M.; Lupu, A.; Ivanov, A.V.; Miron, I.C.; et al. Pitfalls of Thrombotic Microangiopathies in Children: Two Case Reports and Literature Review. Diagnostics 2023, 13, 1228. https://doi.org/10.3390/diagnostics13071228
Mocanu A, Bogos RA, Lazaruc TI, Cianga AL, Lupu VV, Ioniuc I, Alecsa M, Lupu A, Ivanov AV, Miron IC, et al. Pitfalls of Thrombotic Microangiopathies in Children: Two Case Reports and Literature Review. Diagnostics. 2023; 13(7):1228. https://doi.org/10.3390/diagnostics13071228
Chicago/Turabian StyleMocanu, Adriana, Roxana Alexandra Bogos, Tudor Ilie Lazaruc, Anca Lavinia Cianga, Vasile Valeriu Lupu, Ileana Ioniuc, Mirabela Alecsa, Ancuta Lupu, Anca Viorica Ivanov, Ingrith Crenguta Miron, and et al. 2023. "Pitfalls of Thrombotic Microangiopathies in Children: Two Case Reports and Literature Review" Diagnostics 13, no. 7: 1228. https://doi.org/10.3390/diagnostics13071228
APA StyleMocanu, A., Bogos, R. A., Lazaruc, T. I., Cianga, A. L., Lupu, V. V., Ioniuc, I., Alecsa, M., Lupu, A., Ivanov, A. V., Miron, I. C., & Starcea, I. M. (2023). Pitfalls of Thrombotic Microangiopathies in Children: Two Case Reports and Literature Review. Diagnostics, 13(7), 1228. https://doi.org/10.3390/diagnostics13071228