
Identification and Validation of T-Cell Exhaustion Signature for Predicting Prognosis and Immune Response in Pancreatic Cancer by Integrated Analysis of Single-Cell and Bulk RNA Sequencing Data


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source and Clinical Information
2.2. Construction of T-Cell Exhaustion Signature
2.3. Validation of T-Cell Exhaustion-Related Risk Model
2.4. Immune Cell Infiltration Estimation
2.5. Immune Subtype Analysis
2.6. Drug Response Prediction
2.7. Immunohistochemistry Staining
2.8. Pan-Cancer scRNA-Seq Analysis of CD8TEX-Related Genes
2.9. Statistical Analysis
3. Results
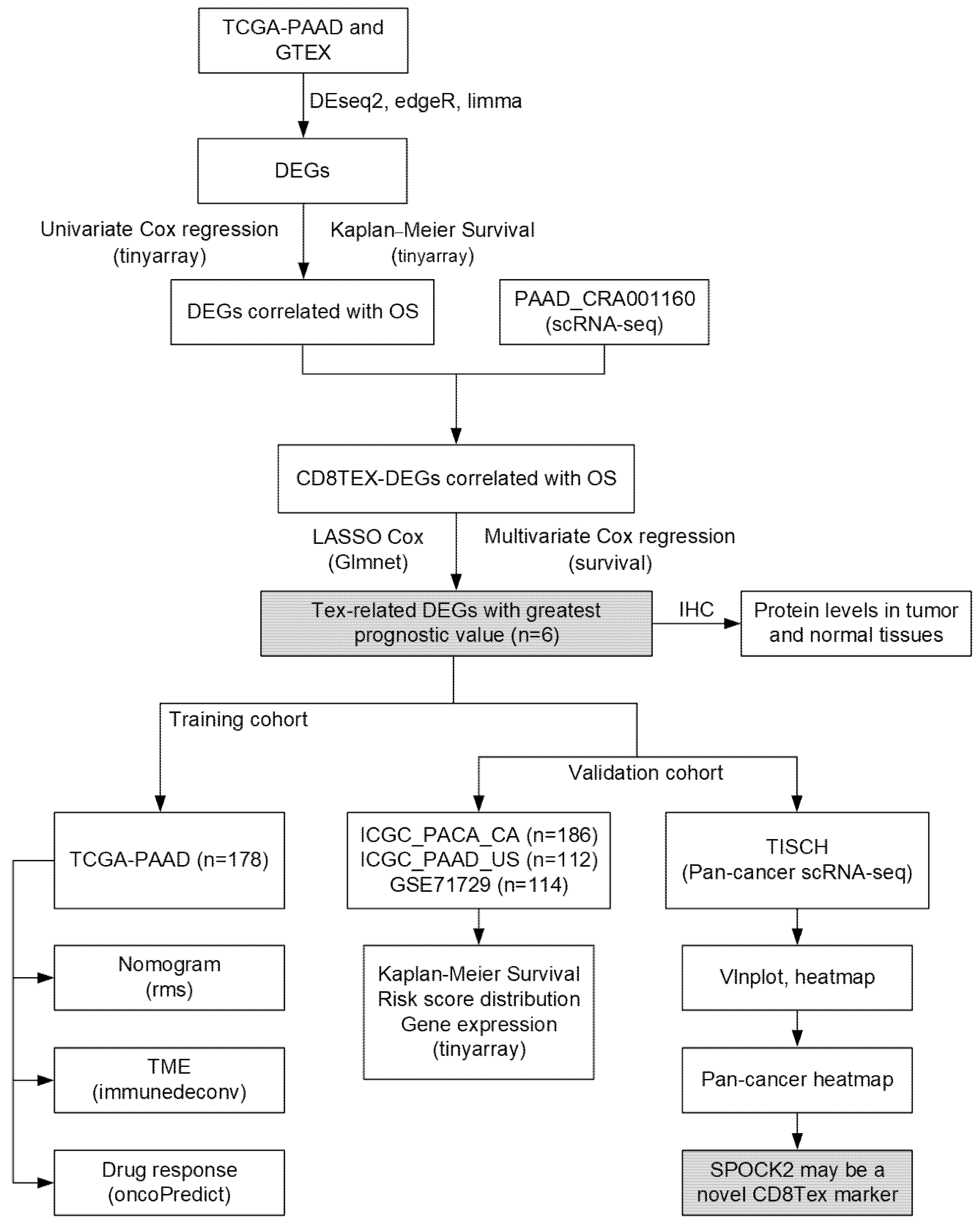
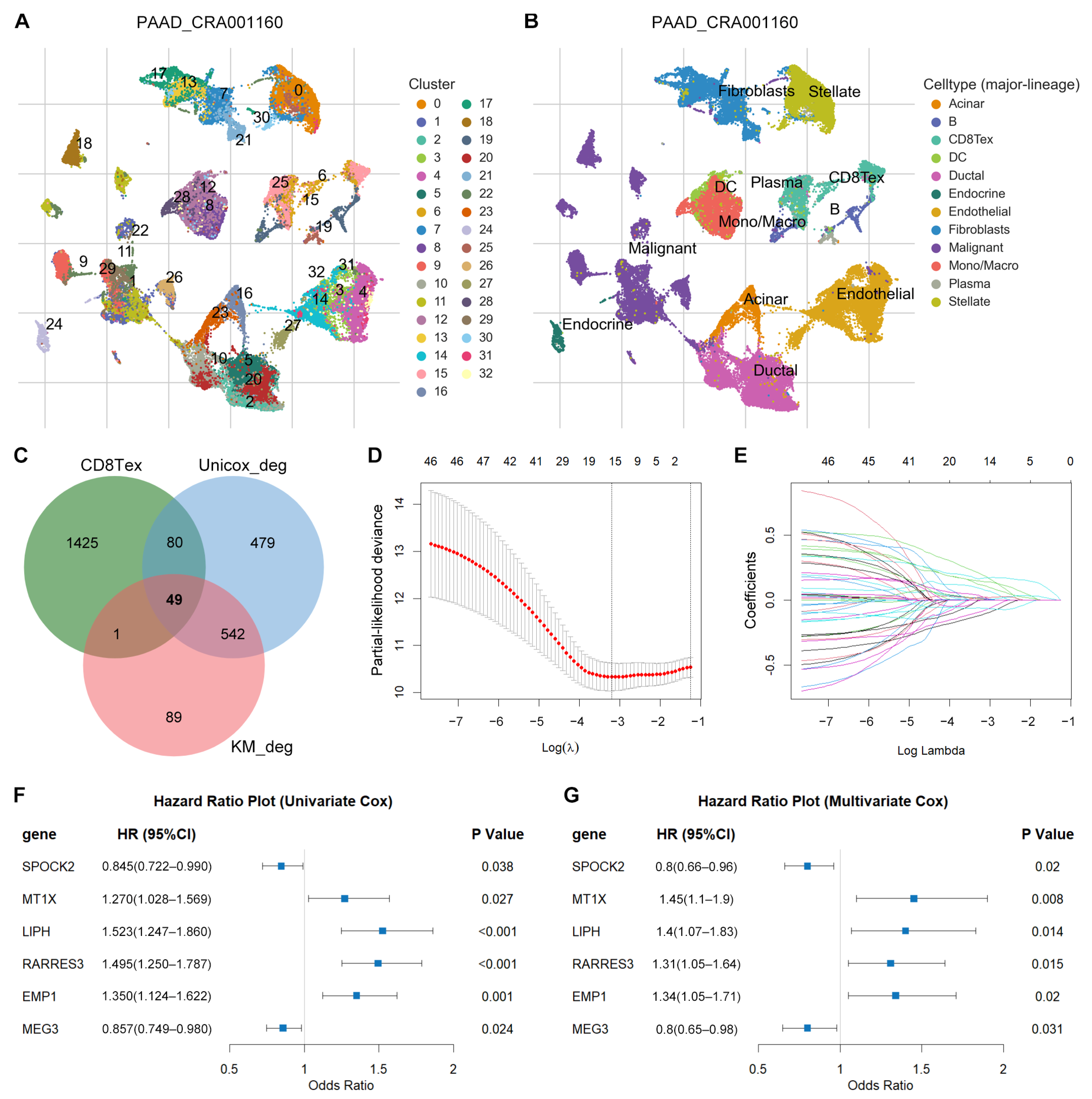
3.1. Identification of TEX-Related Gene Signature in PACA
3.2. Differential Expression Analysis
3.3. Prognostic Potential Analysis of DEGs
3.4. Identification of TEX-Related Gene Signature in PACA
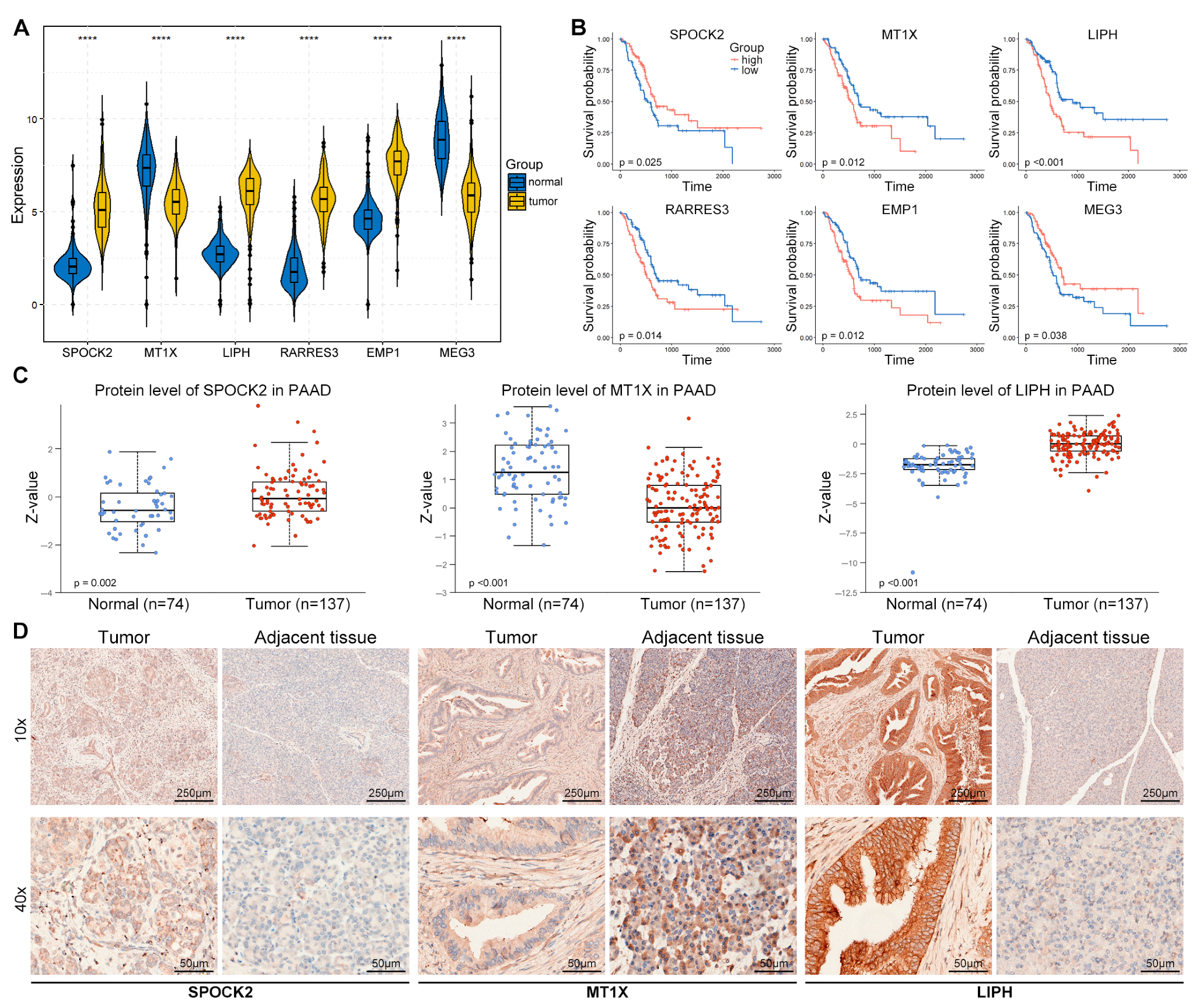
3.5. Validation of TEX-Related Gene Signature in PACA
3.6. Immune Characteristics of High- and Low-Risk Groups
3.7. The Correlation between Drug Sensitivity and Risk Score
3.8. Expression Validation of Risk Genes in PACA Tissues
3.9. SPOCK2 May Be a Novel CD8TEX Cellular Marker
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
| PACA | pancreatic cancer |
| PAAD | pancreatic adenocarcinoma |
| TIME | tumor immune microenvironment |
| TME | tumor microenvironment |
| ICB | immune checkpoint blockade |
| TEX | T-cell exhaustion |
| ScRNA-seq | single-cell RNA sequencing |
| TISCH | Tumor Immune Single-Cell Hub |
| TCGA | Cancer Genome Atlas |
| GTEx | Genotype-Tissue Expression |
| ICGC | International Cancer Genome Consortium |
| GEO | Gene Expression Omnibus |
| DEGs | differentially expressed genes |
| OS | overall survival |
| BCC | basal cell carcinoma |
| LIHC | liver hepatocellular carcinoma |
| PRAD | prostate adenocarcinoma |
| SCC | Squamous cell carcinoma |
| KICH | kidney chromophobe |
| NSCLC | non-small cell lung carcinoma |
| OV | ovarian serous cystadenocarcinoma |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ferlay, J.; Partensky, C.; Bray, F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. 2016, 55, 1158–1160. [Google Scholar] [CrossRef]
- Ilic, M.; Ilic, I. Epidemiology of pancreatic cancer. World J. Gastroenterol. 2016, 22, 9694–9705. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; Koay, E.J.; Chari, S.T.; Maitra, A. Early Detection of Pancreatic Cancer: Opportunities and Challenges. Gastroenterology 2019, 156, 2024–2040. [Google Scholar] [CrossRef] [PubMed]
- Kirkegard, J.; Gaber, C.; Heide-Jorgensen, U.; Fristrup, C.W.; Lund, J.L.; Cronin-Fenton, D.; Mortensen, F.V. Effect of surgery versus chemotherapy in pancreatic cancer patients: A target trial emulation. J. Natl. Cancer Inst. 2024, djae024. [Google Scholar] [CrossRef] [PubMed]
- O'Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab with or Without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Bear, A.S.; Vonderheide, R.H.; O'Hara, M.H. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer Cell 2020, 38, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.Y.; Cao, Y.Z.; Zhou, Y.W.; Ma, Y.S.; Jiang, H.; Zhang, H.; Jiang, L.; Yang, Q.X.; Tang, X.M.; Yang, C.; et al. The power and the promise of CAR-mediated cell immunotherapy for clinical application in pancreatic cancer. J. Adv. Res. 2024, in press. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Baessler, A.; Vignali, D.A.A. T Cell Exhaustion. Annu. Rev. Immunol. 2024, 42. [Google Scholar] [CrossRef]
- Kallies, A.; Zehn, D.; Utzschneider, D.T. Precursor exhausted T cells: Key to successful immunotherapy? Nat. Rev. Immunol. 2020, 20, 128–136. [Google Scholar] [CrossRef]
- Shin, H.; Wherry, E.J. CD8 T cell dysfunction during chronic viral infection. Curr. Opin. Immunol. 2007, 19, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, S.I.; Thommen, D.S.; Moersig, W.; Muller, P.; Zippelius, A. Cancer immunology—Development of novel anti-cancer therapies. Swiss Med. Wkly 2015, 145, w14066. [Google Scholar] [CrossRef]
- Shin, H.; Blackburn, S.D.; Intlekofer, A.M.; Kao, C.; Angelosanto, J.M.; Reiner, S.L.; Wherry, E.J. A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity 2009, 31, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Workman, C.J.; Vignali, D.A.A. LAG-3 as the third checkpoint inhibitor. Nat. Immunol. 2023, 24, 1415–1422. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zeng, G.; Zhou, S.; He, X.; Sun, N.; Zhu, X.; Hu, A. Blocking Tim-3 or/and PD-1 reverses dysfunction of tumor-infiltrating lymphocytes in HBV-related hepatocellular carcinoma. Bull. Cancer 2018, 105, 493–501. [Google Scholar] [CrossRef]
- Huang, B.; Liu, R.; Wang, P.; Yuan, Z.; Yang, J.; Xiong, H.; Zhang, N.; Huang, Q.; Fu, X.; Sun, W.; et al. CD8(+)CD57(+) T cells exhibit distinct features in human non-small cell lung cancer. J. Immunother. Cancer 2020, 8, e000639. [Google Scholar] [CrossRef]
- Guan, Q.; Han, M.; Guo, Q.; Yan, F.; Wang, M.; Ning, Q.; Xi, D. Strategies to reinvigorate exhausted CD8(+) T cells in tumor microenvironment. Front. Immunol. 2023, 14, 1204363. [Google Scholar] [CrossRef]
- Blanche, P.; Dartigues, J.F.; Jacqmin-Gadda, H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat. Med. 2013, 32, 5381–5397. [Google Scholar] [CrossRef]
- He, Y.; Jiang, Z.; Chen, C.; Wang, X. Classification of triple-negative breast cancers based on Immunogenomic profiling. J. Exp. Clin. Cancer Res. 2018, 37, 327. [Google Scholar] [CrossRef]
- Sturm, G.; Finotello, F.; List, M. Immunedeconv: An R Package for Unified Access to Computational Methods for Estimating Immune Cell Fractions from Bulk RNA-Sequencing Data. Methods Mol. Biol. 2020, 2120, 223–232. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830 e814. [Google Scholar] [CrossRef]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief Bioinform. 2021, 22, bbab260. [Google Scholar] [CrossRef]
- Wang, C.; Sun, D.; Huang, X.; Wan, C.; Li, Z.; Han, Y.; Qin, Q.; Fan, J.; Qiu, X.; Xie, Y.; et al. Integrative analyses of single-cell transcriptome and regulome using MAESTRO. Genome Biol. 2020, 21, 198. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, F.; Chandrashekar, D.S.; Varambally, S.; Creighton, C.J. Proteogenomic characterization of 2002 human cancers reveals pan-cancer molecular subtypes and associated pathways. Nat. Commun. 2022, 13, 2669. [Google Scholar] [CrossRef]
- Bai, X.; Li, Y.; Zeng, X.; Zhao, Q.; Zhang, Z. Single-cell sequencing technology in tumor research. Clin. Chim. Acta 2021, 518, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Hsu, C.; Chan, S.L.; Choo, S.P.; Kudo, M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J. Hepatol. 2020, 72, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yin, R.; Xu, L. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 379, e14. [Google Scholar] [CrossRef]
- Blando, J.; Sharma, A.; Higa, M.G.; Zhao, H.; Vence, L.; Yadav, S.S.; Kim, J.; Sepulveda, A.M.; Sharp, M.; Maitra, A.; et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 1692–1697. [Google Scholar] [CrossRef]
- Moncada, R.; Barkley, D.; Wagner, F.; Chiodin, M.; Devlin, J.C.; Baron, M.; Hajdu, C.H.; Simeone, D.M.; Yanai, I. Author Correction: Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 2020, 38, 1476. [Google Scholar] [CrossRef]
- Kuang, T.; Zhang, L.; Chai, D.; Chen, C.; Wang, W. Construction of a T-cell exhaustion-related gene signature for predicting prognosis and immune response in hepatocellular carcinoma. Aging 2023, 15, 5751–5774. [Google Scholar] [CrossRef]
- Zhu, Y.P.; Deng, H.T.; Wang, X.; Rahat, M.A.; Sun, S.; Zhang, Q.Z. Cuproptosis-related molecular subtypes direct T cell exhaustion phenotypes and therapeutic strategies for patients with lung adenocarcinoma. Front. Pharmacol. 2023, 14, 1146468. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Zhao, S.; Yang, J.; Gao, X.; Peng, G.; Zhang, J.; Xie, X.; Song, G.; Xu, K.; Xia, Z.; et al. T-cell exhaustion signatures characterize the immune landscape and predict HCC prognosis via integrating single-cell RNA-seq and bulk RNA-sequencing. Front. Immunol. 2023, 14, 1137025. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, Q.; Tu, K.; Chen, Y.; Peng, Y.; Ni, Y.; Zhu, G.; Cheng, C.; Li, Y.; Xiao, X.; et al. Single-Cell Transcriptomics of Immune Cells Reveal Diversity and Exhaustion Signatures in Non-Small-Cell Lung Cancer. Front. Immunol. 2022, 13, 854724. [Google Scholar] [CrossRef] [PubMed]
- Budimir, N.; Thomas, G.D.; Dolina, J.S.; Salek-Ardakani, S. Reversing T-cell Exhaustion in Cancer: Lessons Learned from PD-1/PD-L1 Immune Checkpoint Blockade. Cancer Immunol. Res. 2022, 10, 146–153. [Google Scholar] [CrossRef]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef] [PubMed]
- Dangaj, D.; Bruand, M.; Grimm, A.J.; Ronet, C.; Barras, D.; Duttagupta, P.A.; Lanitis, E.; Duraiswamy, J.; Tanyi, J.L.; Benencia, F.; et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 2019, 35, 885–900 e810. [Google Scholar] [CrossRef] [PubMed]
- Valpione, S.; Galvani, E.; Tweedy, J.; Mundra, P.A.; Banyard, A.; Middlehurst, P.; Barry, J.; Mills, S.; Salih, Z.; Weightman, J.; et al. Immune-awakening revealed by peripheral T cell dynamics after one cycle of immunotherapy. Nat Cancer 2020, 1, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Fairfax, B.P.; Taylor, C.A.; Watson, R.A.; Nassiri, I.; Danielli, S.; Fang, H.; Mahe, E.A.; Cooper, R.; Woodcock, V.; Traill, Z.; et al. Peripheral CD8(+) T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nat. Med. 2020, 26, 193–199. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef]
- Li, J.; Yuan, S.; Norgard, R.J.; Yan, F.; Sun, Y.H.; Kim, I.K.; Merrell, A.J.; Sela, Y.; Jiang, Y.; Bhanu, N.V.; et al. Epigenetic and Transcriptional Control of the Epidermal Growth Factor Receptor Regulates the Tumor Immune Microenvironment in Pancreatic Cancer. Cancer Discov. 2021, 11, 736–753. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.J.H.; Tang, A.S.P.; Lim, W.H.; Ng, C.H.; Nah, B.; Fu, C.; Xiao, J.; Koh, B.; Tay, P.W.L.; Tan, E.X.; et al. Survival Trends in Sorafenib for Advanced Hepatocellular Carcinoma: A Reconstructed Individual Patient Data Meta-Analysis of Randomized Trials. Liver Cancer 2023, 12, 445–456. [Google Scholar] [CrossRef]
- Christmas, B.J.; Rafie, C.I.; Hopkins, A.C.; Scott, B.A.; Ma, H.S.; Cruz, K.A.; Woolman, S.; Armstrong, T.D.; Connolly, R.M.; Azad, N.A.; et al. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol. Res. 2018, 6, 1561–1577. [Google Scholar] [CrossRef]
- Zhao, J.; Cheng, M.; Gai, J.; Zhang, R.; Du, T.; Li, Q. SPOCK2 Serves as a Potential Prognostic Marker and Correlates with Immune Infiltration in Lung Adenocarcinoma. Front. Genet. 2020, 11, 588499. [Google Scholar] [CrossRef]
- Masiulionyte, B.; Valiulyte, I.; Tamasauskas, A.; Skiriute, D. Metallothionein Genes are Highly Expressed in Malignant Astrocytomas and Associated with Patient Survival. Sci. Rep. 2019, 9, 5406. [Google Scholar] [CrossRef]
- Morales, M.; Arenas, E.J.; Urosevic, J.; Guiu, M.; Fernandez, E.; Planet, E.; Fenwick, R.B.; Fernandez-Ruiz, S.; Salvatella, X.; Reverter, D.; et al. RARRES3 suppresses breast cancer lung metastasis by regulating adhesion and differentiation. EMBO Mol. Med. 2014, 6, 865–881. [Google Scholar] [CrossRef]
- Lu, B.; Wei, J.; Zhou, H.; Chen, J.; Li, Y.; Ye, L.; Zhao, W.; Wu, S. Histone H3K36me2 demethylase KDM2A promotes bladder cancer progression through epigenetically silencing RARRES3. Cell Death Dis. 2022, 13, 547. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.S.; Majami, A.A.; Ali, H.; Gupta, G.; Almalki, W.H.; Alzarea, S.I.; Kazmi, I.; Syed, R.U.; Khalifa, N.E.; Bin Break, M.K.; et al. The complex role of MEG3: An emerging long non-coding RNA in breast cancer. Pathol. Res. Pract. 2023, 251, 154850. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Tan, L.; Luo, D.; Wang, X. Identification and Validation of T-Cell Exhaustion Signature for Predicting Prognosis and Immune Response in Pancreatic Cancer by Integrated Analysis of Single-Cell and Bulk RNA Sequencing Data. Diagnostics 2024, 14, 667. https://doi.org/10.3390/diagnostics14060667
Zhu Y, Tan L, Luo D, Wang X. Identification and Validation of T-Cell Exhaustion Signature for Predicting Prognosis and Immune Response in Pancreatic Cancer by Integrated Analysis of Single-Cell and Bulk RNA Sequencing Data. Diagnostics. 2024; 14(6):667. https://doi.org/10.3390/diagnostics14060667
Chicago/Turabian StyleZhu, Yaowu, Li Tan, Danju Luo, and Xiong Wang. 2024. "Identification and Validation of T-Cell Exhaustion Signature for Predicting Prognosis and Immune Response in Pancreatic Cancer by Integrated Analysis of Single-Cell and Bulk RNA Sequencing Data" Diagnostics 14, no. 6: 667. https://doi.org/10.3390/diagnostics14060667
APA StyleZhu, Y., Tan, L., Luo, D., & Wang, X. (2024). Identification and Validation of T-Cell Exhaustion Signature for Predicting Prognosis and Immune Response in Pancreatic Cancer by Integrated Analysis of Single-Cell and Bulk RNA Sequencing Data. Diagnostics, 14(6), 667. https://doi.org/10.3390/diagnostics14060667