
Intestinal Dysbiosis in Young Cystic Fibrosis Rabbits


 and
and
Abstract
:1. Introduction
2. Results
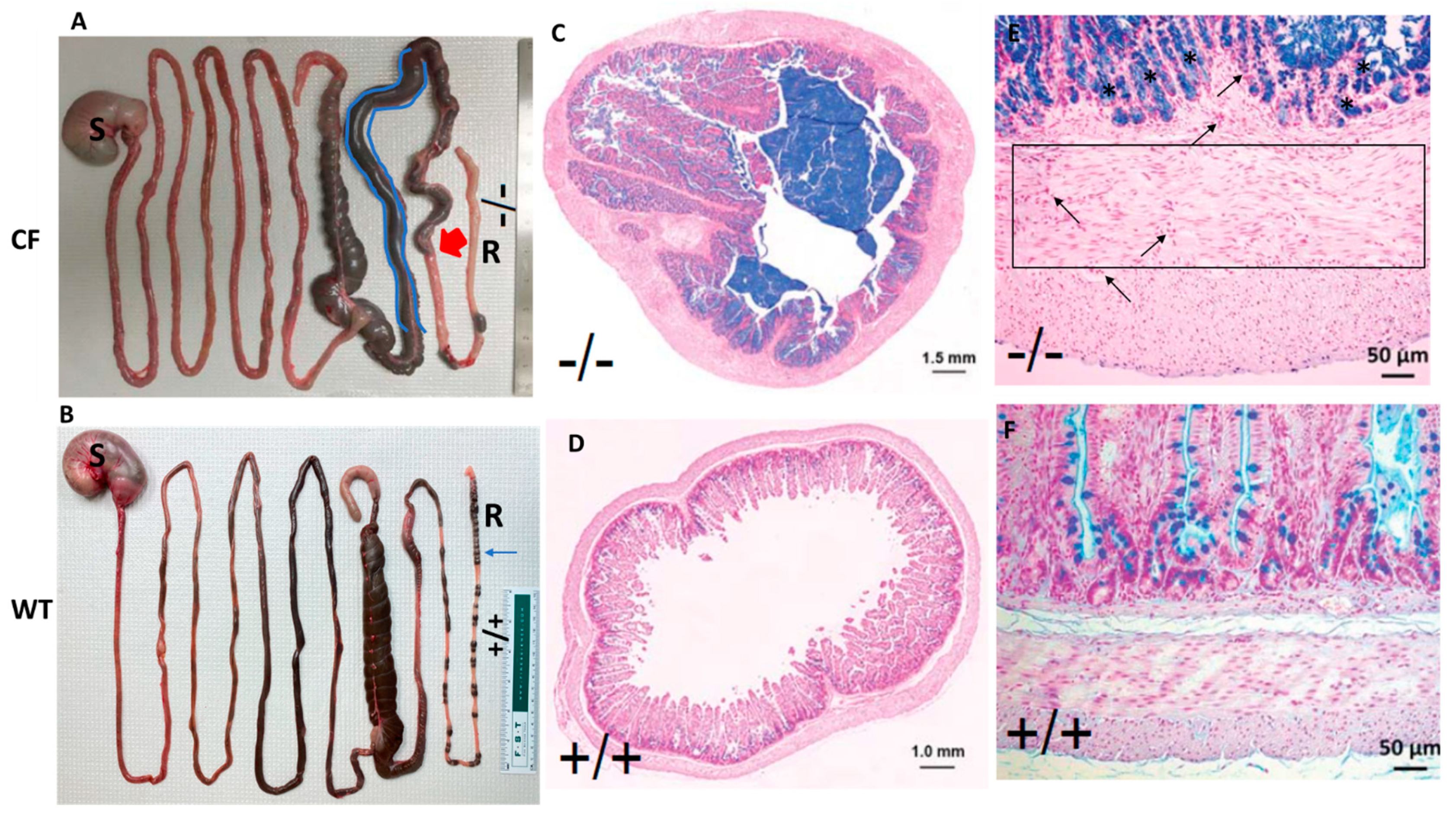
2.1. CF Rabbits Exhibit Intestinal Obstruction
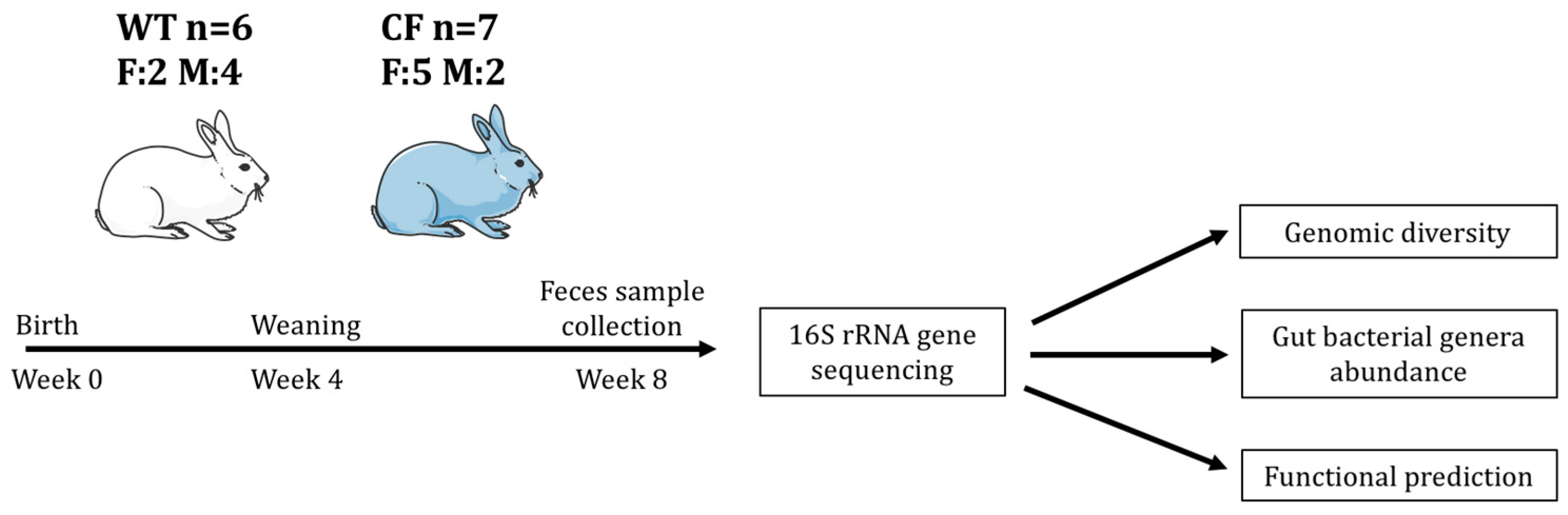
2.2. Study Sample Characteristics
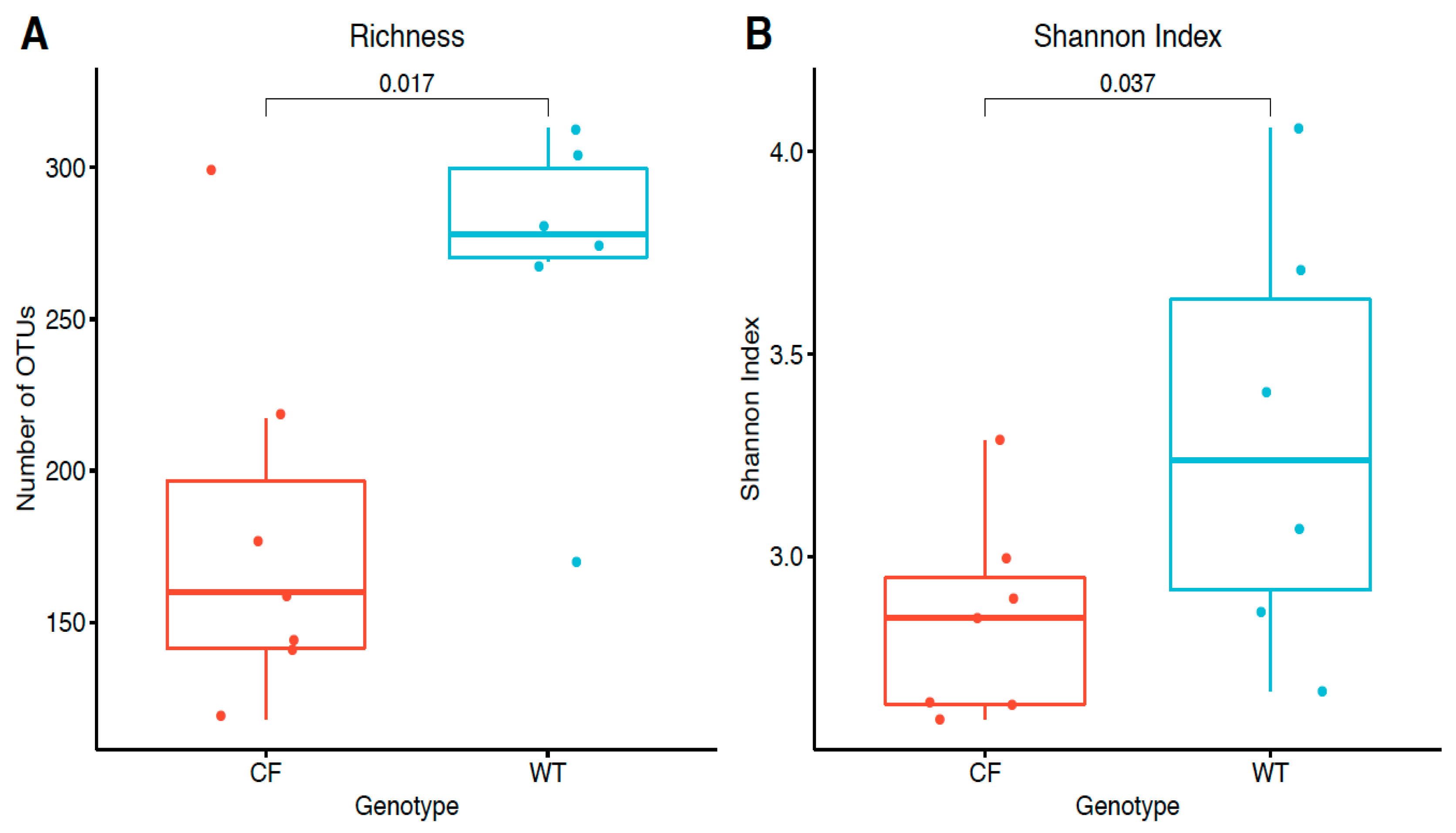
2.3. Alpha Diversity
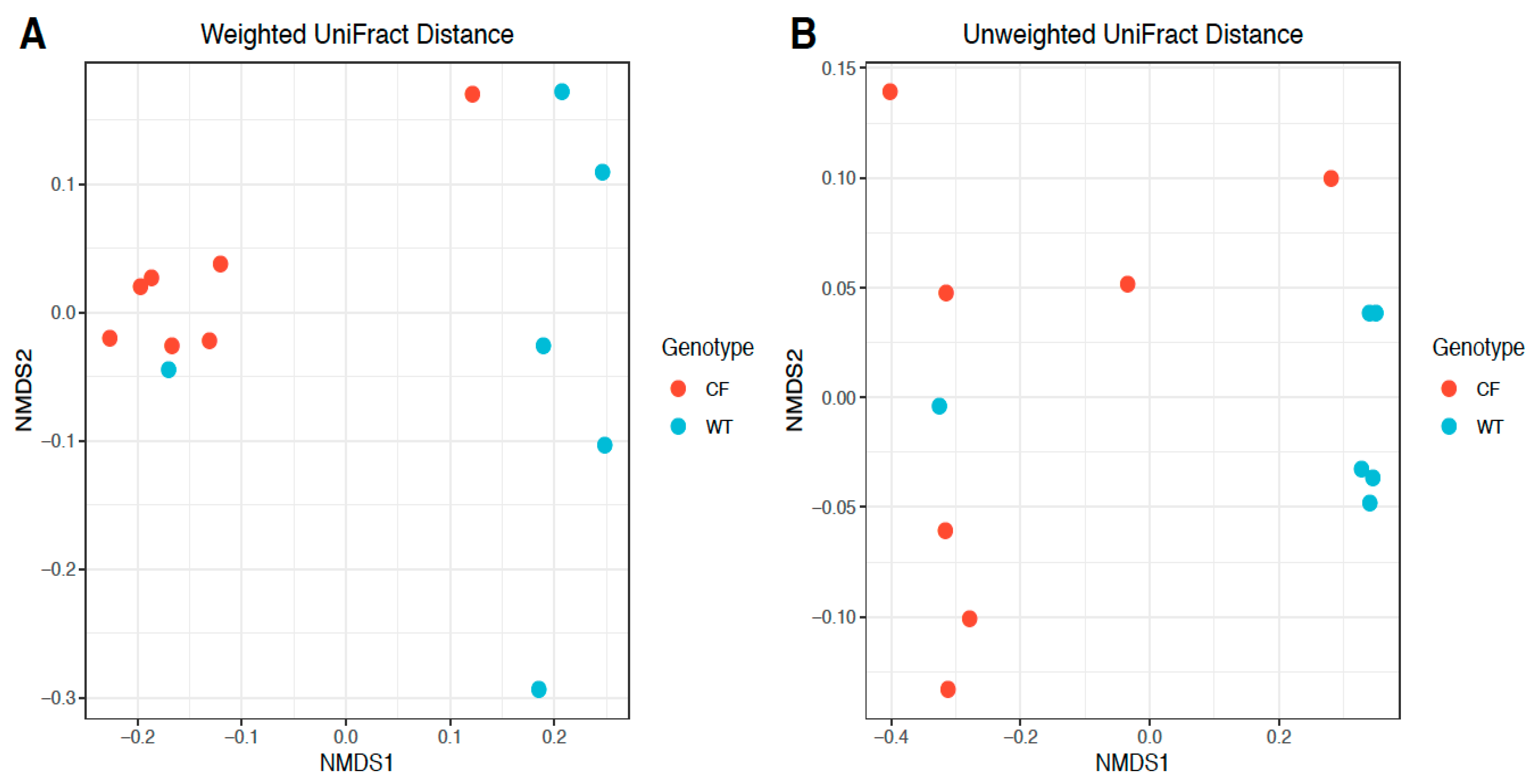
2.4. Beta Diversity
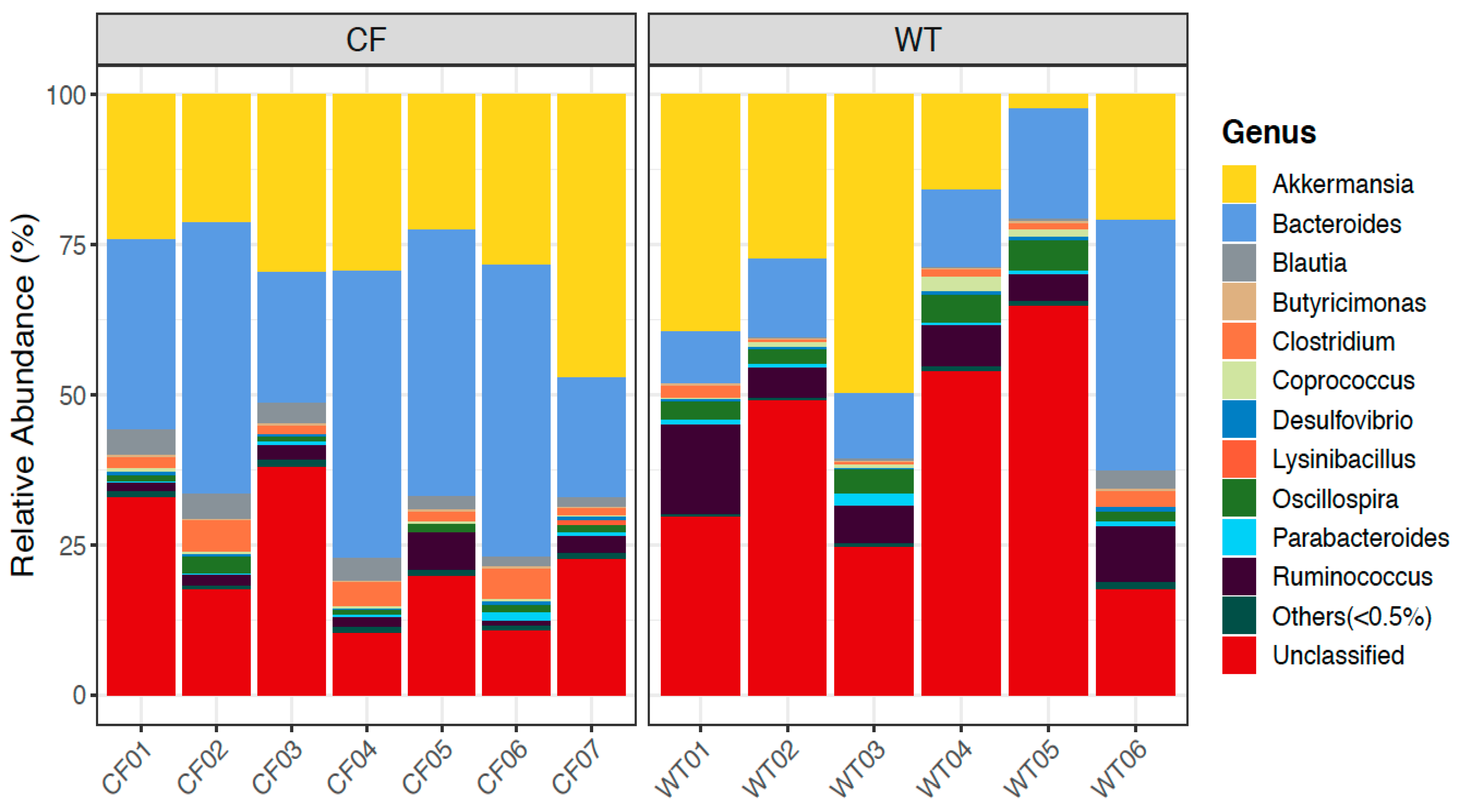
2.5. Relative Abundance of Bacterial Genera in CF and WT Rabbits
2.6. Predicted Functional Analysis by PICRUSt
3. Discussion
4. Materials and Methods
4.1. Animals and Fecal Sample Collection
4.2. Histology Staining
4.3. 16S rRNA Sequencing Data Processing
4.4. Alpha Diversity Analysis
4.5. Beta Diversity Analysis
4.6. Differential Abundance Analysis
4.7. Functional Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Wrennall, J.A.; Cai, Z.; Li, H.; Sheppard, D.N. Understanding how cystic fibrosis mutations disrupt CFTR function: From single molecules to animal models. Int. J. Biochem. Cell Biol. 2014, 52, 47–57. [Google Scholar] [CrossRef]
- Stoltz, D.A.; Meyerholz, D.K.; Welsh, M.J. Origins of cystic fibrosis lung disease. New Engl. J. Med. 2015, 372, 351–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bear, C.E. A Therapy for Most with Cystic Fibrosis. Cell 2020, 180, 211. [Google Scholar] [CrossRef] [PubMed]
- Cystic fibrosis. Nat. Rev. Dis Primers 2015, 1, 15049. [CrossRef] [PubMed]
- Sathe, M.; Houwen, R. Meconium ileus in Cystic Fibrosis. J. Cyst. Fibros. 2017, 16 (Suppl. 2), S32–S39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galante, G.; Freeman, A.J. Gastrointestinal, Pancreatic, and Hepatic Manifestations of Cystic Fibrosis in the Newborn. Neoreviews 2019, 20, e12–e24. [Google Scholar] [CrossRef]
- Van der Doef, H.P.; Kokke, F.T.; van der Ent, C.K.; Houwen, R.H. Intestinal obstruction syndromes in cystic fibrosis: Meconium ileus, distal intestinal obstruction syndrome, and constipation. Curr. Gastroenterol. Rep. 2011, 13, 265–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadjiliadis, D.; Khoruts, A.; Zauber, A.G.; Hempstead, S.E.; Maisonneuve, P.; Lowenfels, A.B. Cystic Fibrosis Colorectal Cancer Screening Task Force. Cystic Fibrosis Colorectal Cancer Screening Consensus Recommendations. Gastroenterology 2018, 154, 736–745.e714. [Google Scholar] [CrossRef] [Green Version]
- Miragoli, F.; Federici, S.; Ferrari, S.; Minuti, A.; Rebecchi, A.; Bruzzese, E.; Buccigrossi, V.; Guarino, A.; Callegari, M.L. Impact of cystic fibrosis disease on archaea and bacteria composition of gut microbiota. FEMS Microbiol. Ecol. 2017, 93, fiw230. [Google Scholar] [CrossRef] [Green Version]
- Antosca, K.M.; Chernikova, D.A.; Price, C.E.; Ruoff, K.L.; Li, K.; Guill, M.F.; Sontag, N.R.; Morrison, H.G.; Hao, S.; Drumm, M.L.; et al. Altered Stool Microbiota of Infants with Cystic Fibrosis Shows a Reduction in Genera Associated with Immune Programming from Birth. J. Bacteriol. 2019, 201, e00274-19. [Google Scholar] [CrossRef] [Green Version]
- Coffey, M.J.; Nielsen, S.; Wemheuer, B.; Kaakoush, N.O.; Garg, M.; Needham, B.; Pickford, R.; Jaffe, A.; Thomas, T.; Ooi, C.Y. Gut Microbiota in Children with Cystic Fibrosis: A Taxonomic and Functional Dysbiosis. Sci. Rep. 2019, 9, 18593. [Google Scholar] [CrossRef]
- Xu, J.; Livraghi-Butrico, A.; Hou, X.; Rajagopalan, C.; Zhang, J.; Song, J.; Jiang, H.; Wei, H.G.; Wang, H.; Bouhamdan, M.; et al. Phenotypes of CF rabbits generated by CRISPR/Cas9-mediated disruption of the CFTR gene. JCI Insight 2021, 6, e139813. [Google Scholar]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [Green Version]
- Cox, T.F.; Cox, M.A.A. Multidimensional Scaling, 2nd ed.; Chapman & Hall/CRC: Boca Raton, FL, USA, 2001. [Google Scholar]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Goldfarb, K.C.; Wild, Y.K.; Kong, W.; De Lisle, R.C.; Brodie, E.L. Cystic fibrosis transmembrane conductance regulator knockout mice exhibit aberrant gastrointestinal microbiota. Gut Microbes 2013, 4, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Bazett, M.; Honeyman, L.; Stefanov, A.N.; Pope, C.E.; Hoffman, L.R.; Haston, C.K. Cystic fibrosis mouse model-dependent intestinal structure and gut microbiome. Mamm. Genome 2015, 26, 222–234. [Google Scholar] [PubMed] [Green Version]
- Meeker, S.M.; Mears, K.S.; Sangwan, N.; Brittnacher, M.J.; Weiss, E.J.; Treuting, P.M.; Tolley, N.; Pope, C.E.; Hager, K.R.; Vo, A.T.; et al. CFTR dysregulation drives active selection of the gut microbiome. Plos Pathog 2020, 16, e1008251. [Google Scholar] [CrossRef]
- Sun, X.; Olivier, A.K.; Yi, Y.; Pope, C.E.; Hayden, H.S.; Liang, B.; Sui, H.; Zhou, W.; Hager, K.R.; Zhang, Y.; et al. Gastrointestinal pathology in juvenile and adult CFTR-knockout ferrets. Am. J. Pathol. 2014, 184, 1309–1322. [Google Scholar] [CrossRef] [Green Version]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Rietschel, E.T. Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 2001, 7, 167–202. [Google Scholar] [CrossRef] [PubMed]
- Trinh, N.T.; Bilodeau, C.; Maillé, E.; Ruffin, M.; Quintal, M.-C.; Desrosiers, M.-Y.; Rousseau, S.; Brochiero, E. Deleterious impact of Pseudomonas aeruginosa on cystic fibrosis transmembrane conductance regulator function and rescue in airway epithelial cells. Eur. Respir. J. 2015, 45, 1590–1602. [Google Scholar] [CrossRef] [Green Version]
- Stanton, B.A.; Coutermarsh, B.; Barnaby, R.; Hogan, D. Pseudomonas aeruginosa Reduces VX-809 Stimulated F508del-CFTR Chloride Secretion by Airway Epithelial Cells. PLoS ONE 2015, 10, e0127742. [Google Scholar] [CrossRef]
- Laselva, O.; Stone, T.A.; Bear, C.E.; Deber, C.M. Anti-Infectives Restore ORKAMBI((R)) Rescue of F508del-CFTR Function in Human Bronchial Epithelial Cells Infected with Clinical Strains of P. aeruginosa. Biomolecules 2020, 10, 334. [Google Scholar]
- Verhelst, X.; Dias, A.M.; Colombel, J.F.; Vermeire, S.; Van Vlierberghe, H.; Callewaert, N.; Pinho, S.S. Protein Glycosylation as a Diagnostic and Prognostic Marker of Chronic Inflammatory Gastrointestinal and Liver Diseases. Gastroenterology 2020, 158, 95–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomme, B.; van Steenkiste, C.; Callewaert, N.; van Vlierberghe, H. Alteration of protein glycosylation in liver diseases. J. Hepatol. 2009, 50, 592–603. [Google Scholar] [PubMed]
- Mollica, M.P.; Mattace Raso, G.; Cavaliere, G.; Trinchese, G.; De Filippo, C.; Aceto, S.; Prisco, M.; Pirozzi, C.; Di Guida, F.; Lama, A.; et al. Butyrate Regulates Liver Mitochondrial Function, Efficiency, and Dynamics in Insulin-Resistant Obese Mice. Diabetes 2017, 66, 1405–1418. [Google Scholar] [CrossRef] [Green Version]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Q.; Jin, G.; Wang, G.; Liu, T.; Liu, X.; Wang, B.; Cao, H. Current Sampling Methods for Gut Microbiota: A Call for More Precise Devices. Front. Cell Infect. Microbiol. 2020, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- Fadrosh, D.W.; Ma, B.; Gajer, P.; Sengamalay, N.; Ott, S.; Brotman, R.M.; Ravel, J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2014, 2, 6. [Google Scholar] [CrossRef] [Green Version]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar]
- Anderson, M.J. A New Method for Non-parametric Multivariate Analysis of Variance. Austral. Ecol. 2001, 26, 32–46. [Google Scholar]
- White, R.; Nagarajan, N.; Pop, M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Markowitz, V.M.; Chen, I.A.; Palaniappan, K.; Chu, K.; Szeto, E.; Grechkin, Y.; Ratner, A.; Jacob, B.; Huang, J.; Williams, P.; et al. IMG: The Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res. 2012, 40, D115–D122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Genotype | Age (days) | Sex | # Tags/sample | # OTUs/sample |
|---|---|---|---|---|---|
| CF01 | CF | 61 | F | 33230 | 217 |
| CF02 | CF | 61 | F | 33029 | 144 |
| CF03 | CF | 53 | F | 35122 | 176 |
| CF04 | CF | 59 | F | 34490 | 139 |
| CF05 | CF | 55 | M | 33458 | 160 |
| CF06 | CF | 53 | M | 34617 | 118 |
| CF07 | CF | 55 | F | 35189 | 299 |
| WT01 | WT | 51 | M | 31635 | 269 |
| WT02 | WT | 51 | F | 29621 | 281 |
| WT03 | WT | 54 | M | 32713 | 275 |
| WT04 | WT | 54 | M | 30499 | 306 |
| WT05 | WT | 53 | F | 29410 | 313 |
| WT06 | WT | 51 | M | 34495 | 170 |
| Taxon | CF Relative Abundance (%) (mean ± SD) | WT Relative Abundance (%) (mean ± SD) | Mean Difference (%) (CF − WT) | p-Value | FDR |
|---|---|---|---|---|---|
| Phylum | |||||
| Firmicutes | 24.234 ± 3.975 | 41.307 ± 11.565 | −17.073 | 0.002 | 0.023 |
| Saccharibacteria | 0.001 ± 0.003 | 0.081 ± 0.075 | −0.08 | 0.010 | 0.035 |
| Bacteroidetes | 45.023 ± 10.483 | 27.355 ± 11.337 | 17.669 | 0.035 | 0.087 |
| Cyanobacteria | 0.008 ± 0.012 | 0.071 ± 0.076 | −0.063 | 0.047 | 0.095 |
| Genus | |||||
| Bacteroides | 37.115 ± 12.404 | 17.673 ± 12.282 | 19.442 | 0.008 | 0.060 |
| Blautia | 2.997 ± 1.147 | 0.67 ± 1.118 | 2.327 | 0.008 | 0.060 |
| Oscillospira | 1.392 ± 0.718 | 3.517 ± 1.347 | −2.125 | 0.005 | 0.060 |
| Roseburia | 0 ± 0 | 0.02 ± 0.023 | −0.02 | 0.006 | 0.060 |
| Ruminococcus | 2.384 ± 1.836 | 7.777 ± 3.881 | −5.392 | 0.008 | 0.060 |
| Holdemania | 0.06 ± 0.04 | 0.015 ± 0.015 | 0.045 | 0.014 | 0.086 |
| Dehalobacterium | 0.012 ± 0.029 | 0.08 ± 0.063 | −0.069 | 0.017 | 0.089 |
| KEGG Orthology | CF Relative Abundance (%) (mean ± SD) | WT Relative Abundance (%) (mean ± SD) | Mean Difference (%) (CF − WT) | p-Value | FDR |
|---|---|---|---|---|---|
| Aminoacyl-tRNA biosynthesis | 1.065 ± 0.032 | 1.117 ± 0.029 | −0.053 | 0.002 | 0.056 |
| Arginine and proline metabolism | 1.361 ± 0.007 | 1.321 ± 0.041 | 0.041 | 0.005 | 0.056 |
| Bacterial chemotaxis | 0.304 ± 0.053 | 0.427 ± 0.102 | −0.123 | 0.008 | 0.056 |
| Chaperones and folding catalysts | 1.05 ± 0.015 | 0.989 ± 0.042 | 0.061 | 0.005 | 0.056 |
| Glycine, serine and threonine metabolism | 0.873 ± 0.013 | 0.832 ± 0.025 | 0.041 | 0.005 | 0.056 |
| Lipid biosynthesis proteins | 0.634 ± 0.018 | 0.68 ± 0.025 | −0.046 | 0.008 | 0.056 |
| Membrane and intracellular structural molecules | 0.712 ± 0.051 | 0.573 ± 0.088 | 0.14 | 0.008 | 0.056 |
| Other glycan degradation | 0.487 ± 0.052 | 0.356 ± 0.078 | 0.131 | 0.008 | 0.056 |
| Propanoate metabolism | 0.495 ± 0.017 | 0.568 ± 0.048 | −0.073 | 0.008 | 0.056 |
| Purine metabolism | 2.062 ± 0.033 | 1.976 ± 0.028 | 0.086 | 0.001 | 0.056 |
| Sporulation | 0.439 ± 0.051 | 0.639 ± 0.159 | −0.2 | 0.008 | 0.056 |
| Bacterial motility proteins | 0.728 ± 0.148 | 1.016 ± 0.198 | −0.287 | 0.014 | 0.071 |
| Butanoate metabolism | 0.588 ± 0.022 | 0.661 ± 0.067 | −0.074 | 0.014 | 0.071 |
| Lipopolysaccharide biosynthesis proteins | 0.549 ± 0.036 | 0.43 ± 0.099 | 0.119 | 0.014 | 0.071 |
| RNA degradation | 0.496 ± 0.01 | 0.456 ± 0.032 | 0.041 | 0.014 | 0.071 |
| Flagellar assembly | 0.224 ± 0.083 | 0.374 ± 0.13 | −0.15 | 0.022 | 0.093 |
| Mismatch repair | 0.748 ± 0.017 | 0.781 ± 0.032 | −0.033 | 0.022 | 0.093 |
| Transcription factors | 1.46 ± 0.049 | 1.609 ± 0.144 | −0.149 | 0.022 | 0.093 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, X.; Bouhamdan, M.; Hou, X.; Zhang, K.; Song, J.; Hao, K.; Jin, J.-P.; Zhang, Z.; Xu, J. Intestinal Dysbiosis in Young Cystic Fibrosis Rabbits. J. Pers. Med. 2021, 11, 132. https://doi.org/10.3390/jpm11020132
Liang X, Bouhamdan M, Hou X, Zhang K, Song J, Hao K, Jin J-P, Zhang Z, Xu J. Intestinal Dysbiosis in Young Cystic Fibrosis Rabbits. Journal of Personalized Medicine. 2021; 11(2):132. https://doi.org/10.3390/jpm11020132
Chicago/Turabian StyleLiang, Xiubin, Mohamad Bouhamdan, Xia Hou, Kezhong Zhang, Jun Song, Ke Hao, Jian-Ping Jin, Zhongyang Zhang, and Jie Xu. 2021. "Intestinal Dysbiosis in Young Cystic Fibrosis Rabbits" Journal of Personalized Medicine 11, no. 2: 132. https://doi.org/10.3390/jpm11020132
APA StyleLiang, X., Bouhamdan, M., Hou, X., Zhang, K., Song, J., Hao, K., Jin, J. -P., Zhang, Z., & Xu, J. (2021). Intestinal Dysbiosis in Young Cystic Fibrosis Rabbits. Journal of Personalized Medicine, 11(2), 132. https://doi.org/10.3390/jpm11020132