
Differential Interactome Proposes Subtype-Specific Biomarkers and Potential Therapeutics in Renal Cell Carcinomas




Abstract
:1. Introduction
2. Materials and Methods
2.1. Collecting of Gene Expression Data
2.2. Obtaining Protein–Protein Interactions Data
2.3. Identification of Differential Interactome and Differentially Interacting Proteins
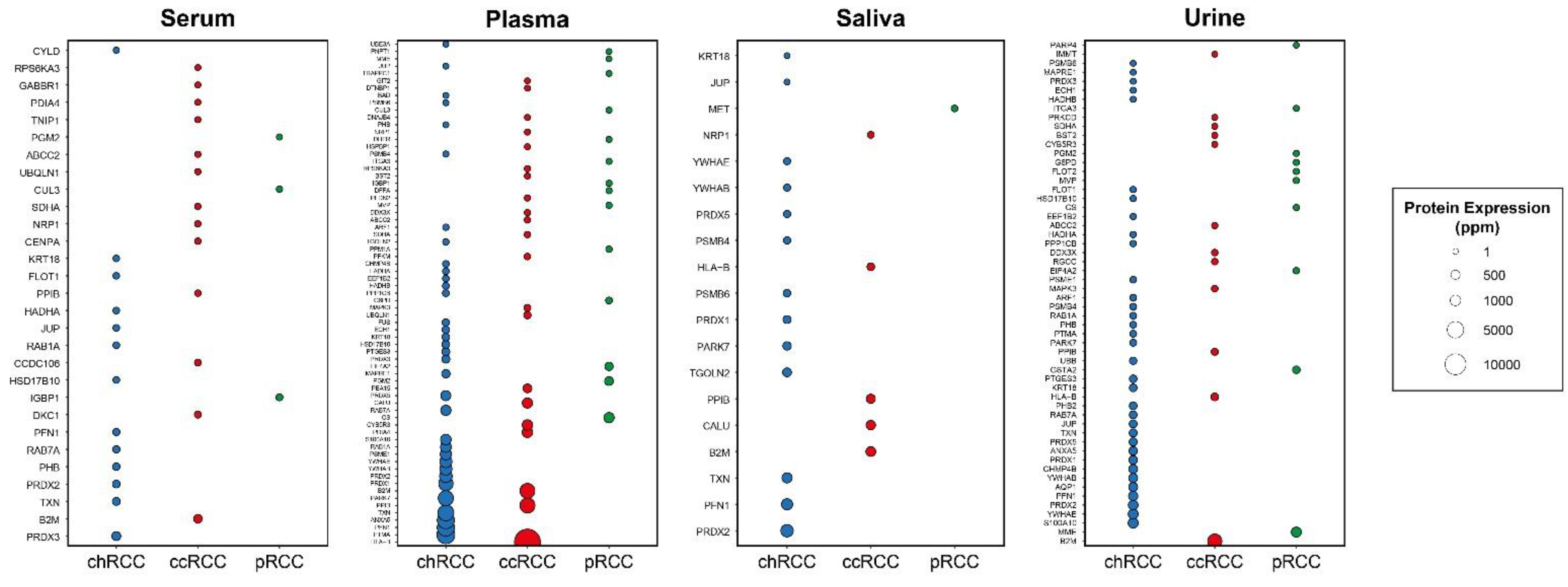
2.4. Evaluation of the Secretion Levels of Subtype-Specific DIPs in Body Fluids
2.5. Analysis of Diagnostic Performance and Prognostic Power
2.6. Identification of Candidate Drugs through Virtual Screening
3. Results
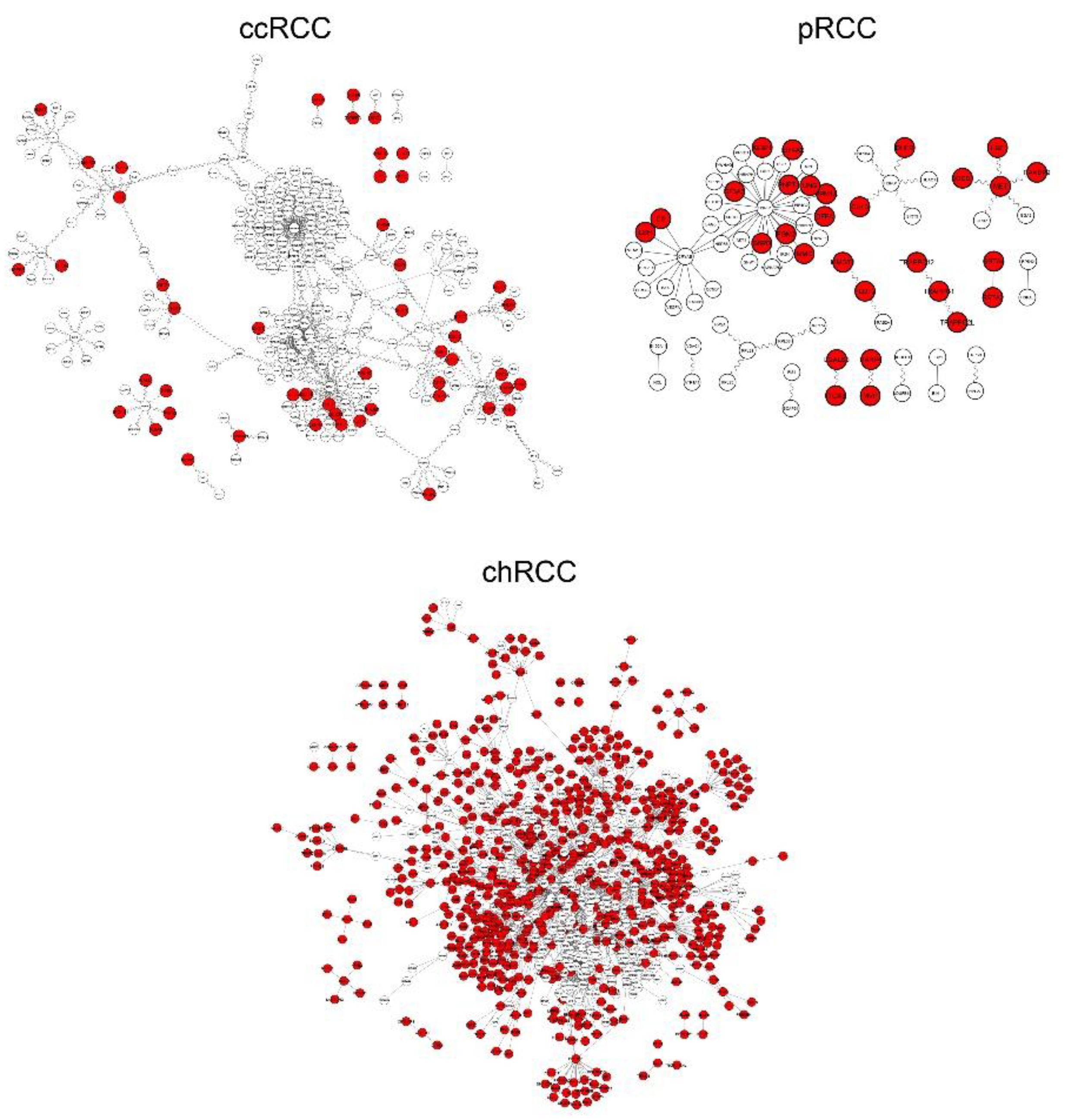
3.1. Differential Interactome Estimation in Subtypes of RCC
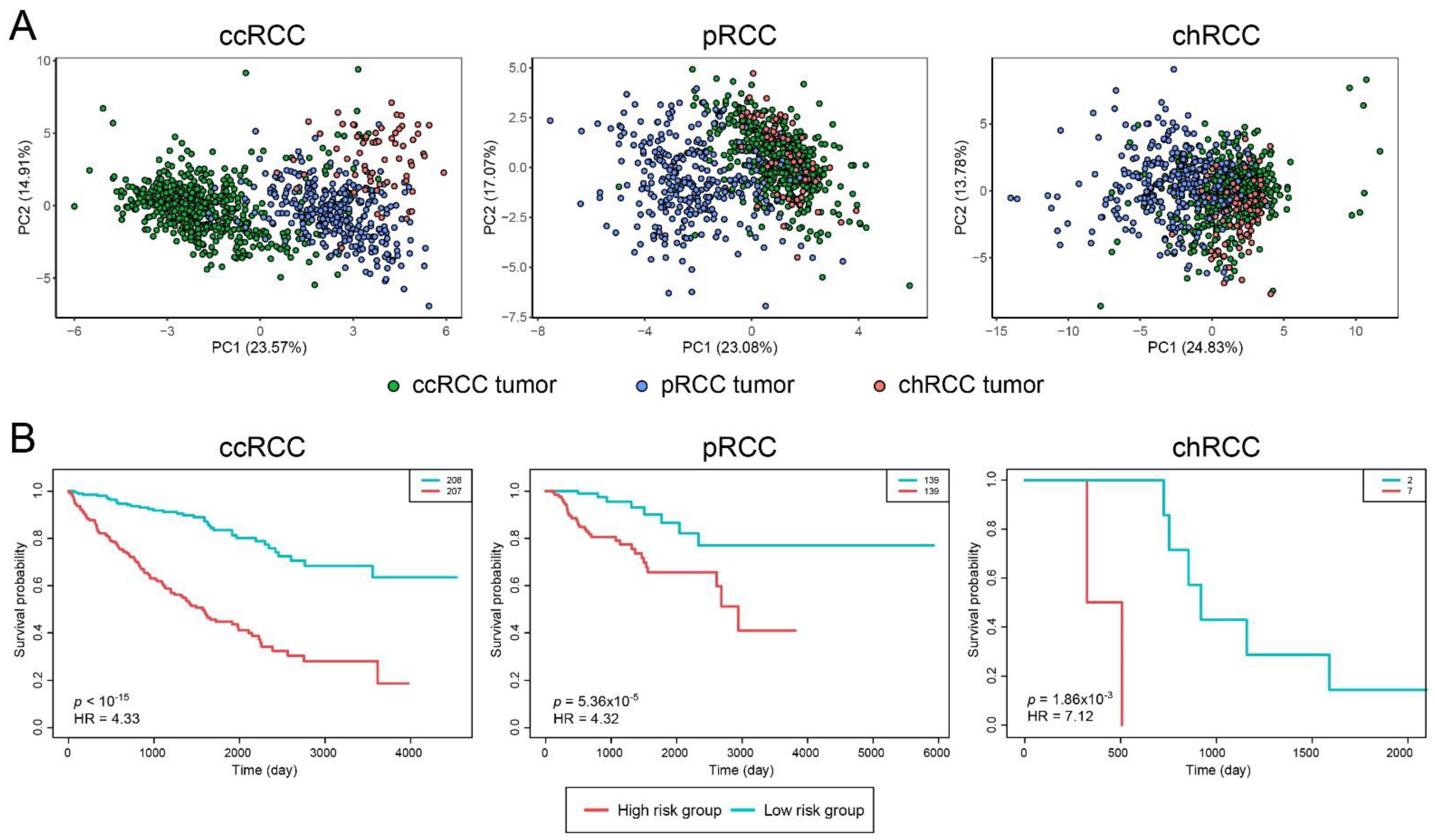
3.2. Prognostic and Diagnostic Capabilities of DIPs Clusters
3.3. Discovery of Drug Candidates through Virtual Screening Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ccRCC | Clear Cell Renal Cell Carcinoma |
| chRCC | Chromophobe Renal Cell Carcinoma |
| DIP | Differentially interacting protein |
| dPPI | Differential protein–protein interaction |
| HR | Hazard ratio |
| KICH | Kidney Chromophobe |
| KIRC | Kidney Renal Clear Cell Carcinoma |
| KIRP | Kidney Renal Papillary Cell Carcinoma |
| LE | Ligand efficiency |
| ns-DIP | Non-secreted DIP |
| PC | Principle component |
| PCA | Principal component analysis |
| PDB | Protein data bank |
| PPI | Protein–protein interaction |
| pRCC | Papillary Renal Cell Carcinoma |
| RCC | Renal cell carcinoma |
| s-DIP | Secreted DIP |
| TCGA | The Cancer Genome Atlas |
| ZINC | ZINC Is Not Commercial |
References
- Barata, P.C.; Rini, B.I. Treatment of renal cell carcinoma: Current status and future directions. CA Cancer J. Clin. 2017, 67, 507–524. [Google Scholar] [CrossRef] [Green Version]
- Linehan, W.M.; Ricketts, C.J. The Cancer Genome Atlas of renal cell carcinoma: Findings and clinical implications. Nat. Rev. Urol. 2019, 16, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Priolo, C.; Khabibullin, D.; Reznik, E.; Filippakis, H.; Ogórek, B.; Kavanagh, T.R.; Nijmeh, J.; Herbert, Z.T.; Asara, J.M.; Kwiatkowski, D.J. Impairment of gamma-glutamyl transferase 1 activity in the metabolic pathogenesis of chromophobe renal cell carcinoma. Proc. Natl. Acad. Sci. USA 2018, 115, E6274–E6282. [Google Scholar] [CrossRef] [Green Version]
- Cairns, P. Renal cell carcinoma. Cancer Biomark. 2011, 9, 461–473. [Google Scholar] [CrossRef]
- Petejova, N.; Martinek, A. Renal cell carcinoma: Review of etiology, pathophysiology and risk factors. Biomed. Pap. Med. Fac. Palacky Univ. Olomouc 2016, 160, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Su, J.; Fu, D.; Wang, Y.; Chen, Y.; Chen, R.; Qin, G.; Zuo, J.; Yue, D. The role of YB1 in renal cell carcinoma cell adhesion. Int. J. Med. Sci. 2018, 15, 1304. [Google Scholar] [CrossRef] [Green Version]
- Caliskan, A.; Andac, A.C.; Arga, K.Y. Novel molecular signatures and potential therapeutics in renal cell carcinomas: Insights from a comparative analysis of subtypes. Genomics 2020, 112, 3166–3178. [Google Scholar] [CrossRef]
- Gulfidan, G.; Turanli, B.; Beklen, H.; Sinha, R.; Arga, K.Y. Pan-cancer mapping of differential protein-protein interactions. Sci. Rep. 2020, 10, 3272. [Google Scholar] [CrossRef]
- Ayyildiz, D.; Gov, E.; Sinha, R.; Arga, K.Y. Ovarian Cancer Differential Interactome and Network Entropy Analysis Reveal New Candidate Biomarkers. Omi. A J. Integr. Biol. 2017, 21, 285–294. [Google Scholar] [CrossRef]
- Turanli, B.; Karagoz, K.; Bidkhori, G.; Sinha, R.; Gatza, M.L.; Uhlen, M.; Mardinoglu, A.; Arga, K.Y. Multi-omic data interpretation to repurpose subtype specific drug candidates for breast cancer. Front. Genet. 2019, 10, 420. [Google Scholar] [CrossRef] [Green Version]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Chatr-Aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A.; et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017, 45, D369–D379. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Iny Stein, T.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Samaras, P.; Schmidt, T.; Frejno, M.; Gessulat, S.; Reinecke, M.; Jarzab, A.; Zecha, J.; Mergner, J.; Giansanti, P.; Ehrlich, H.-C.; et al. ProteomicsDB: A multi-omics and multi-organism resource for life science research. Nucleic Acids Res. 2019, 48, D1153–D1163. [Google Scholar] [CrossRef] [PubMed]
- Schaab, C.; Geiger, T.; Stoehr, G.; Cox, J.; Mann, M. Analysis of high accuracy, quantitative proteomics data in the MaxQB database. Mol. Cell. Proteom. 2012, 11, M111.014068. [Google Scholar] [CrossRef] [Green Version]
- Montague, E.; Stanberry, L.; Higdon, R.; Janko, I.; Lee, E.; Anderson, N.; Choiniere, J.; Stewart, E.; Yandl, G.; Broomall, W.; et al. MOPED 2.5—An integrated multi-omics resource: Multi-omics profiling expression database now includes transcriptomics data. Omics J. Integr. Biol. 2014, 18, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martínez-Ledesma, E.; Martínez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Peña, J.G.; Treviño, V. SurvExpress: An Online Biomarker Validation Tool and Database for Cancer Gene Expression Data Using Survival Analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchanan, S.G.; Hendle, J.; Lee, P.S.; Smith, C.R.; Bounaud, P.-Y.; Jessen, K.A.; Tang, C.M.; Huser, N.H.; Felce, J.D.; Froning, K.J. SGX523 is an exquisitely selective, ATP-competitive inhibitor of the MET receptor tyrosine kinase with antitumor activity in vivo. Mol. Cancer Ther. 2009, 8, 3181–3190. [Google Scholar] [CrossRef] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Chem. Biol. 2015, 1263, 243–250. [Google Scholar]
- Jonsson, P.F.; Bates, P.A. Global topological features of cancer proteins in the human interactome. Bioinformatics 2006, 22, 2291–2297. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: A novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: An important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef]
- Nagaraja, G.M.; Kaur, P.; Asea, A. Role of human and mouse HspB1 in metastasis. Curr. Mol. Med. 2012, 12, 1142–1150. [Google Scholar] [CrossRef]
- Ljungberg, B.; Cowan, N.C.; Hanbury, D.C.; Hora, M.; Kuczyk, M.A.; Merseburger, A.S.; Patard, J.-J.; Mulders, P.F.A.; Sinescu, I.C. EAU guidelines on renal cell carcinoma: The 2010 update. Eur. Urol. 2010, 58, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Wu, Z.Z.; Xu, Y.; Wang, J.G.; Wang, Y.Q.; Cao, M.Q.; Wang, C.H. Saliva proteomic analysis reveals possible biomarkers of renal cell carcinoma. Open Chem. 2020, 18, 918–926. [Google Scholar] [CrossRef]
- Dudek, A.; Appleyard, L.; O’Brien, T.S.; Chowdhury, S.; Champion, P.; Challacombe, B.; Kooiman, G.; Carpenter, G. Salivary markers in renal cell carcinoma. J. Clin. Oncol. 2014, 32, 489. [Google Scholar] [CrossRef]
- Ryu, J.W.; Han, S.-Y.; Yun, J.I.; Choi, S.-U.; Jung, H.; Du Ha, J.; Cho, S.Y.; Lee, C.O.; Kang, N.S.; Koh, J.S. Design and synthesis of triazolopyridazines substituted with methylisoquinolinone as selective c-Met kinase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 7185–7188. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specificity | s-DIPs 1 | Non s-DIPs 2 |
|---|---|---|
| ccRCC-specific | ABCC2, B2M, BST2, CALU, CCDC106, CENPA, CYB5R3, DDX3X, DKC1, DNAJB4, DTNBP1, GABBR1, GIT2, HLA-B, HSPBP1, IMMT, MAPK3, NRP1, PDIA4, PEA15, PFDN2, PFKM, PPIB, PRKCD, RGCC, RPS6KA3, SDHA, UBQLN1, TNIP1 | AZIN1, CDT1, ELF4, FBXW8, GPS2, IL32, IRF1, LDOC1, MCM7, MCM9, MTF1, MTOR, P4HA2, PHLPP1, RSL1D1, SCD, TAF1, TAPBP, TOMM20, USP2, ZNF668 |
| pRCC-specific | CS, CUL3, DFFA, DHFR, EIF4A2, FLOT2, G6PD, GSTA2, IGBP1, ITGA3, MET, MME, MVP, PARP4, PGM2, PNPT1, PPM1A, TRAPPC1 | GSTA4, HGF, LBH, LGALS8, MMGT1, RANBP9, SF3A3, SOCS1, TRAPPC12, TRAPPC2L, UNG |
| chRCC-specific | ANXA5, AQP1, ARF1, BAD, CHMP4B, CYLD, ECH1, EEF1B2, FLOT1, FUS, HADHA, HADHB, HSD17B10, JUP, KRT18, MAPRE1, PARK7, PFN1, PHB, PHB2, PPP1CB, PRDX1, PRDX2, PRDX3, PRDX5, PSMB4, PSMB6, PSME1, PTGES3, PTMA, RAB1A, RAB7A, S100A10, TGOLN2, TXN, UBB, UBE3A, YWHAB, YWHAE | ABL1, AMFR, ARAF, CDK9, FOS, JUND, MCL1, MORF4L2, SF3B5, STAU1, TRIM8 |
| Ligand ZINC15 ID | Vina Binding Affinity (kcal/mol) | Ligand Efficiency (LE) |
|---|---|---|
| ZINC200458361 | −12.7 | 0.41 |
| ZINC144529139 | −12.6 | 0.39 |
| ZINC73196087 | −12.6 | 0.45 |
| ZINC72318117 | −12.5 | 0.44 |
| ZINC72318118 | −12.5 | 0.41 |
| ZINC73163075 | −12.5 | 0.42 |
| ZINC96284612 | −12.5 | 0.41 |
| ZINC150080371 | −12.4 | 0.38 |
| ZINC299865209 | −12.4 | 0.42 |
| ZINC43176957 | −12.4 | 0.43 |
| ZINC73165724 | −12.4 | 0.39 |
| ZINC73196196 | −12.4 | 0.43 |
| ZINC72318119 | −12.3 | 0.41 |
| ZINC150078084 | −12.2 | 0.37 |
| ZINC96284613 | −12.2 | 0.39 |
| ZINC144475075 | −12.1 | 0.4 |
| ZINC40431067 | −12.1 | 0.37 |
| ZINC84759584 | −12.1 | 0.36 |
| ZINC96284618 | −12.1 | 0.37 |
| ZINC144529348 | −12 | 0.41 |
| ZINC166085169 | −12 | 0.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caliskan, A.; Gulfidan, G.; Sinha, R.; Arga, K.Y. Differential Interactome Proposes Subtype-Specific Biomarkers and Potential Therapeutics in Renal Cell Carcinomas. J. Pers. Med. 2021, 11, 158. https://doi.org/10.3390/jpm11020158
Caliskan A, Gulfidan G, Sinha R, Arga KY. Differential Interactome Proposes Subtype-Specific Biomarkers and Potential Therapeutics in Renal Cell Carcinomas. Journal of Personalized Medicine. 2021; 11(2):158. https://doi.org/10.3390/jpm11020158
Chicago/Turabian StyleCaliskan, Aysegul, Gizem Gulfidan, Raghu Sinha, and Kazim Yalcin Arga. 2021. "Differential Interactome Proposes Subtype-Specific Biomarkers and Potential Therapeutics in Renal Cell Carcinomas" Journal of Personalized Medicine 11, no. 2: 158. https://doi.org/10.3390/jpm11020158
APA StyleCaliskan, A., Gulfidan, G., Sinha, R., & Arga, K. Y. (2021). Differential Interactome Proposes Subtype-Specific Biomarkers and Potential Therapeutics in Renal Cell Carcinomas. Journal of Personalized Medicine, 11(2), 158. https://doi.org/10.3390/jpm11020158