
Aberrant Gamma-Band Oscillations in Mice with Vitamin D Deficiency: Implications on Schizophrenia and its Cognitive Symptoms


Abstract
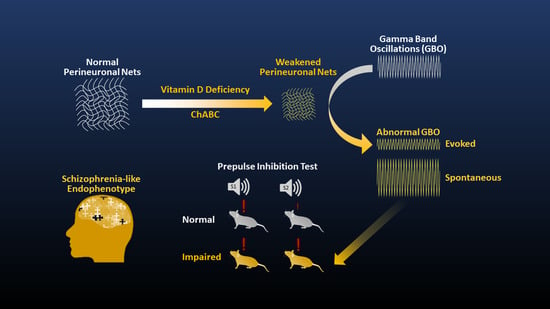
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal
2.2. Stereotaxic Surgery
2.3. Experimental Design
2.4. Optogenetically-Evoked GBO
2.5. Auditory Steady-State Response (ASSR)
2.6. In Vivo Electrophysiology Data Acquisition
2.7. Chondroitinase ABC Treatment
2.8. Immunohistochemistry
2.9. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McNally, J.M.; McCarley, R.W. Gamma band oscillations: A key to understanding schizophrenia symptoms and neural circuit abnormalities. Curr. Opin. Psychiatry 2016, 29, 202–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, O.; Kaiser, J.; Lachaux, J.P. Human gamma-frequency oscillations associated with attention and memory. Trends Neurosci. 2007, 30, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Gregoriou, G.G.; Gotts, S.J.; Zhou, H.; Desimone, R. High-frequency, long-range coupling between prefrontal and visual cortex during attention. Science 2009, 324, 1207–1210. [Google Scholar] [CrossRef] [Green Version]
- Rouhinen, S.; Panula, J.; Palva, J.M.; Palva, S. Load dependence of beta and gamma oscillations predicts individual capacity of visual attention. J. Neurosci. 2013, 33, 19023–19033. [Google Scholar] [CrossRef] [Green Version]
- Colgin, L.L.; Denninger, T.; Fyhn, M.; Hafting, T.; Bonnevie, T.; Jensen, O.; Moser, M.B.; Moser, E.I. Frequency of gamma oscillations routes flow of information in the hippocampus. Nature 2009, 462, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Nakazono, T.; Jun, H.; Blurton-Jones, M.; Green, K.N.; Igarashi, K.M. Gamma oscillations in the entorhinal-hippocampal circuit underlying memory and dementia. Neurosci. Res. 2018, 129, 40–46. [Google Scholar] [CrossRef]
- Uhlhaas, P.J.; Singer, W. Abnormal neural oscillations and synchrony in schizophrenia. Nat. Rev. Neurosci. 2010, 11, 100–113. [Google Scholar] [CrossRef]
- Koenig, T.; Prichep, L.; Dierks, T.; Hubl, D.; Wahlund, L.O.; John, E.R.; Jelic, V. Decreased EEG synchronization in Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging. 2005, 26, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Ethridge, L.E.; White, S.P.; Mosconi, M.W.; Wang, J.; Byerly, M.J.; Sweeney, J.A. Reduced habituation of auditory evoked potentials indicate cortical hyper-excitability in Fragile X Syndrome. Transl. Psychiatry 2016, 6, e787. [Google Scholar] [CrossRef]
- Adaikkan, C.; Tsai, L.H. Gamma entrainment: Impact on neurocircuits, glia, and therapeutic opportunities. Trends Neurosci. 2020, 43, 24–41. [Google Scholar] [CrossRef]
- Buzsaki, G.; Wang, X.J. Mechanisms of gamma oscillations. Annu. Rev. Neurosci. 2012, 35, 203–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vreugdenhil, M.; Jefferys, J.G.; Celio, M.R.; Schwaller, B. Parvalbumin-deficiency facilitates repetitive IPSCs and gamma oscillations in the hippocampus. J. Neurophysiol. 2003, 89, 1414–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.; Thankachan, S.; McKenna, J.T.; McNally, J.M.; Yang, C.; Choi, J.H.; Chen, L.; Kocsis, B.; Deisseroth, K.; Strecker, R.E.; et al. Cortically projecting basal forebrain parvalbumin neurons regulate cortical gamma band oscillations. Proc. Natl. Acad. Sci. USA 2015, 112, 3535–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohal, V.S.; Zhang, F.; Yizhar, O.; Deisseroth, K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 2009, 459, 698–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepnicki, P.; Kondej, M.; Kaczor, A.A. Current concepts and treatments of schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, R.S.; Keefe, R.S. Schizophrenia is a cognitive illness: Time for a change in focus. JAMA Psychiatry 2013, 70, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.K.; Hoch, R.; Lee, A.T.; Patel, T.; Rubenstein, J.L.; Sohal, V.S. Gamma rhythms link prefrontal interneuron dysfunction with cognitive inflexibility in Dlx5/6(+/−) mice. Neuron 2015, 85, 1332–1343. [Google Scholar] [CrossRef] [Green Version]
- Cadinu, D.; Grayson, B.; Podda, G.; Harte, M.K.; Doostdar, N.; Neill, J.C. NMDA receptor antagonist rodent models for cognition in schizophrenia and identification of novel drug treatments, an update. Neuropharmacology 2018, 142, 41–62. [Google Scholar] [CrossRef] [Green Version]
- Sultana, R.; Brooks, C.B.; Shrestha, A.; Ogundele, O.M.; Lee, C.C. Perineuronal nets in the prefrontal cortex of a schizophrenia mouse model: Assessment of neuroanatomical, electrophysiological, and behavioral contributions. Int. J. Mol. Sci. 2021, 22, 11140. [Google Scholar] [CrossRef]
- Sun, Y.; Farzan, F.; Barr, M.S.; Kirihara, K.; Fitzgerald, P.B.; Light, G.A.; Daskalakis, Z.J. gamma oscillations in schizophrenia: Mechanisms and clinical significance. Brain Res. 2011, 1413, 98–114. [Google Scholar] [CrossRef]
- Kwon, J.S.; O’Donnell, B.F.; Wallenstein, G.V.; Greene, R.W.; Hirayasu, Y.; Nestor, P.G.; Hasselmo, M.E.; Potts, G.F.; Shenton, M.E.; McCarley, R.W. Gamma frequency-range abnormalities to auditory stimulation in schizophrenia. Arch. Gen. Psychiatry 1999, 56, 1001–1005. [Google Scholar] [CrossRef]
- Grutzner, C.; Wibral, M.; Sun, L.; Rivolta, D.; Singer, W.; Maurer, K.; Uhlhaas, P.J. Deficits in high- (>60 Hz) gamma-band oscillations during visual processing in schizophrenia. Front. Hum. Neurosci. 2013, 7, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.; Mellin, J.M.; Alagapan, S.; Alexander, M.L.; Gilmore, J.H.; Jarskog, L.F.; Frohlich, F. Targeting reduced neural oscillations in patients with schizophrenia by transcranial alternating current stimulation. Neuroimage 2019, 186, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Minzenberg, M.J.; Firl, A.J.; Yoon, J.H.; Gomes, G.C.; Reinking, C.; Carter, C.S. Gamma oscillatory power is impaired during cognitive control independent of medication status in first-episode schizophrenia. Neuropsychopharmacology 2010, 35, 2590–2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, K.M. Baseline gamma power during auditory steady-state stimulation in schizophrenia. Front. Hum. Neurosci. 2011, 5, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, Y.; Oribe, N.; Kanba, S.; Onitsuka, T.; Nestor, P.G.; Spencer, K.M. Spontaneous gamma activity in schizophrenia. JAMA Psychiatry 2015, 72, 813–821. [Google Scholar] [CrossRef]
- Sohal, V.S.; Rubenstein, J.L.R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Saunders, J.A.; Gandal, M.J.; Siegel, S.J. NMDA antagonists recreate signal-to-noise ratio and timing perturbations present in schizophrenia. Neurobiol. Dis. 2012, 46, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berretta, S.; Pantazopoulos, H.; Markota, M.; Brown, C.; Batzianouli, E.T. Losing the sugar coating: Potential impact of perineuronal net abnormalities on interneurons in schizophrenia. Schizophr. Res. 2015, 167, 18–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Cacquevel, M.; Saksida, L.M.; Bussey, T.J.; Schneider, B.L.; Aebischer, P.; Melani, R.; Pizzorusso, T.; Fawcett, J.W.; Spillantini, M.G. Perineuronal net digestion with chondroitinase restores memory in mice with tau pathology. Exp. Neurol. 2015, 265, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, G.; Tan, C.L.; Alves, J.N.; Ehlert, E.M.E.; Miller, G.M.; Hsieh-Wilson, L.C.; Sugahara, K.; Oosterhof, A.; van Kuppevelt, T.H.; Verhaagen, J.; et al. Semaphorin 3A binds to the perineuronal nets via chondroitin sulfate type E motifs in rodent brains. J. Biol. Chem. 2013, 288, 27384–27395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, T.H.; Binder, D.K.; Ethell, I.M.; Razak, K.A. The perineuronal ’safety’ net? perineuronal net abnormalities in neurological disorders. Front. Mol. Neurosci. 2018, 11, 270. [Google Scholar] [CrossRef]
- Enwright, J.F.; Sanapala, S.; Foglio, A.; Berry, R.; Fish, K.N.; Lewis, D.A. Reduced labeling of parvalbumin neurons and perineuronal nets in the dorsolateral prefrontal cortex of subjects with schizophrenia. Neuropsychopharmacology 2016, 41, 2206–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauney, S.A.; Athanas, K.M.; Pantazopoulos, H.; Shaskan, N.; Passeri, E.; Berretta, S.; Woo, T.U. Developmental pattern of perineuronal nets in the human prefrontal cortex and their deficit in schizophrenia. Biol. Psychiatry 2013, 74, 427–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcaide, J.; Guirado, R.; Crespo, C.; Blasco-Ibanez, J.M.; Varea, E.; Sanjuan, J.; Nacher, J. Alterations of perineuronal nets in the dorsolateral prefrontal cortex of neuropsychiatric patients. Int. J. Bipolar Disord. 2019, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, R.; Lipachev, N.; Matuszko, G.; Kochneva, A.; Dvoeglazova, A.; Becker, A.; Paveliev, M.; Dityatev, A. Fine structure analysis of perineuronal nets in the ketamine model of schizophrenia. Eur. J. Neurosci. 2021, 53, 3988–4004. [Google Scholar] [CrossRef] [PubMed]
- Tewari, B.P.; Chaunsali, L.; Campbell, S.L.; Patel, D.C.; Goode, A.E.; Sontheimer, H. Perineuronal nets decrease membrane capacitance of peritumoral fast spiking interneurons in a model of epilepsy. Nat. Commun. 2018, 9, 4724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balmer, T.S. Perineuronal nets enhance the excitability of fast-spiking neurons. eNeuro 2016, 3, 745–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingert, J.C.; Sorg, B.A. Impact of perineuronal nets on electrophysiology of parvalbumin interneurons, principal neurons, and brain oscillations: A review. Front. Synaptic Neurosci. 2021, 13, 673210. [Google Scholar] [CrossRef]
- Cui, X.; McGrath, J.J.; Burne, T.H.J.; Eyles, D.W. Vitamin D and schizophrenia: 20 years on. Mol. Psychiatry 2021, 26, 2708–2720. [Google Scholar] [CrossRef] [PubMed]
- Mayne, P.E.; Burne, T.H.J. Vitamin D in synaptic plasticity, cognitive function, and neuropsychiatric illness. Trends Neurosci. 2019, 42, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, P.B.; Pedersen, C.B.; Westergaard, T.; Wohlfahrt, J.; Ewald, H.; Mors, O.; Andersen, P.K.; Melbye, M. Effects of family history and place and season of birth on the risk of schizophrenia. N. Engl. J. Med. 1999, 340, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Eyles, D.W.; Trzaskowski, M.; Vinkhuyzen, A.A.E.; Mattheisen, M.; Meier, S.; Gooch, H.; Anggono, V.; Cui, X.; Tan, M.C.; Burne, T.H.J.; et al. The association between neonatal vitamin D status and risk of schizophrenia. Sci. Rep. 2018, 8, 17692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Amin, M.M.; Sullivan, R.K.P.; Kurniawan, N.D.; Burne, T.H.J. Adult vitamin D deficiency disrupts hippocampal-dependent learning and structural brain connectivity in BALB/c mice. Brain Struct. Funct. 2019, 224, 1315–1329. [Google Scholar] [CrossRef]
- Alaiyed, S.; Bozzelli, P.L.; Caccavano, A.; Wu, J.Y.; Conant, K. Venlafaxine stimulates PNN proteolysis and MMP-9-dependent enhancement of gamma power; relevance to antidepressant efficacy. J. Neurochem. 2019, 148, 810–821. [Google Scholar] [CrossRef]
- Hayani, H.; Song, I.; Dityatev, A. Increased excitability and reduced excitatory synaptic input into fast-spiking CA2 interneurons after enzymatic attenuation of extracellular matrix. Front. Cell Neurosci. 2018, 12, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favuzzi, E.; Marques-Smith, A.; Deogracias, R.; Winterflood, C.M.; Sanchez-Aguilera, A.; Mantoan, L.; Maeso, P.; Fernandes, C.; Ewers, H.; Rico, B. Activity-dependent gating of parvalbumin interneuron function by the perineuronal net protein brevican. Neuron 2017, 95, 639–655.e610. [Google Scholar] [CrossRef] [PubMed]
- Lensjo, K.K.; Lepperod, M.E.; Dick, G.; Hafting, T.; Fyhn, M. Removal of perineuronal nets unlocks juvenile plasticity through network mechanisms of decreased inhibition and increased gamma activity. J. Neurosci. 2017, 37, 1269–1283. [Google Scholar] [CrossRef] [Green Version]
- Mena, A.; Ruiz-Salas, J.C.; Puentes, A.; Dorado, I.; Ruiz-Veguilla, M.; De la Casa, L.G. Reduced Prepulse Inhibition as a Biomarker of Schizophrenia. Front. Behav. Neurosci. 2016, 10, 202. [Google Scholar] [CrossRef] [Green Version]
- Osipova, D.; Pekkonen, E.; Ahveninen, J. Enhanced magnetic auditory steady-state response in early Alzheimer’s disease. Clin. Neurophysiol. 2006, 117, 1990–1995. [Google Scholar] [CrossRef]
- Oda, Y.; Onitsuka, T.; Tsuchimoto, R.; Hirano, S.; Oribe, N.; Ueno, T.; Hirano, Y.; Nakamura, I.; Miura, T.; Kanba, S. Gamma band neural synchronization deficits for auditory steady state responses in bipolar disorder patients. PLoS ONE 2012, 7, e39955. [Google Scholar] [CrossRef] [Green Version]
- Seymour, R.A.; Rippon, G.; Gooding-Williams, G.; Sowman, P.F.; Kessler, K. Reduced auditory steady state responses in autism spectrum disorder. Mol. Autism. 2020, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- van Diessen, E.; Senders, J.; Jansen, F.E.; Boersma, M.; Bruining, H. Increased power of resting-state gamma oscillations in autism spectrum disorder detected by routine electroencephalography. Eur. Arch. Psychiatry Clin. Neurosci. 2015, 265, 537–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dursun, E.; Gezen-Ak, D.; Yilmazer, S. A novel perspective for Alzheimer’s disease: Vitamin D receptor suppression by amyloid-beta and preventing the amyloid-beta induced alterations by vitamin D in cortical neurons. J. Alzheimers Dis. 2011, 23, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Brewer, L.D.; Thibault, V.; Chen, K.C.; Langub, M.C.; Landfield, P.W.; Porter, N.M. Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J. Neurosci. 2001, 21, 98–108. [Google Scholar] [CrossRef]
- Gottschling, C.; Wegrzyn, D.; Denecke, B.; Faissner, A. Elimination of the four extracellular matrix molecules tenascin-C, tenascin-R, brevican and neurocan alters the ratio of excitatory and inhibitory synapses. Sci. Rep. 2019, 9, 13939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, M.R.; Salmen, B.; Bukalo, O.; Rollenhagen, A.; Bosl, M.R.; Morellini, F.; Bartsch, U.; Dityatev, A.; Schachner, M. Impairment of L-type Ca2+ channel-dependent forms of hippocampal synaptic plasticity in mice deficient in the extracellular matrix glycoprotein tenascin-C. J. Neurosci. 2002, 22, 7177–7194. [Google Scholar] [CrossRef] [Green Version]
- Dityatev, A.; Seidenbecher, C.I.; Schachner, M. Compartmentalization from the outside: The extracellular matrix and functional microdomains in the brain. Trends Neurosci. 2010, 33, 503–512. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, S.; Park, M.; Kang, J.; Lee, E.; Jung, J.; Kim, T. Aberrant Gamma-Band Oscillations in Mice with Vitamin D Deficiency: Implications on Schizophrenia and its Cognitive Symptoms. J. Pers. Med. 2022, 12, 318. https://doi.org/10.3390/jpm12020318
Yu S, Park M, Kang J, Lee E, Jung J, Kim T. Aberrant Gamma-Band Oscillations in Mice with Vitamin D Deficiency: Implications on Schizophrenia and its Cognitive Symptoms. Journal of Personalized Medicine. 2022; 12(2):318. https://doi.org/10.3390/jpm12020318
Chicago/Turabian StyleYu, Seungyeong, Mincheol Park, Jiseung Kang, Eunkyung Lee, Jieun Jung, and Tae Kim. 2022. "Aberrant Gamma-Band Oscillations in Mice with Vitamin D Deficiency: Implications on Schizophrenia and its Cognitive Symptoms" Journal of Personalized Medicine 12, no. 2: 318. https://doi.org/10.3390/jpm12020318
APA StyleYu, S., Park, M., Kang, J., Lee, E., Jung, J., & Kim, T. (2022). Aberrant Gamma-Band Oscillations in Mice with Vitamin D Deficiency: Implications on Schizophrenia and its Cognitive Symptoms. Journal of Personalized Medicine, 12(2), 318. https://doi.org/10.3390/jpm12020318