
Personalized Immunotherapies for Type 1 Diabetes: Who, What, When, and How?


Abstract
:1. Introduction
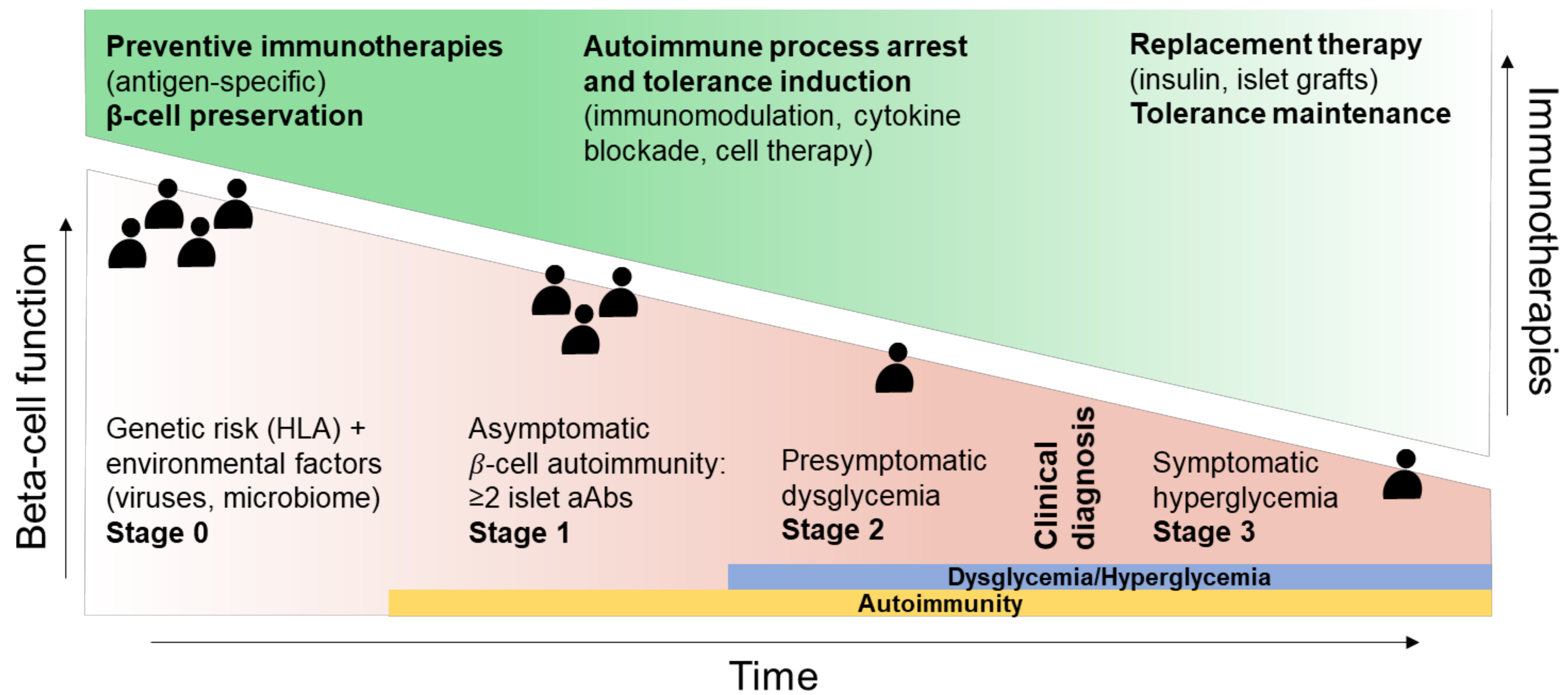
2. Disease Stage (“When”)
3. Endotypes (“Who”)
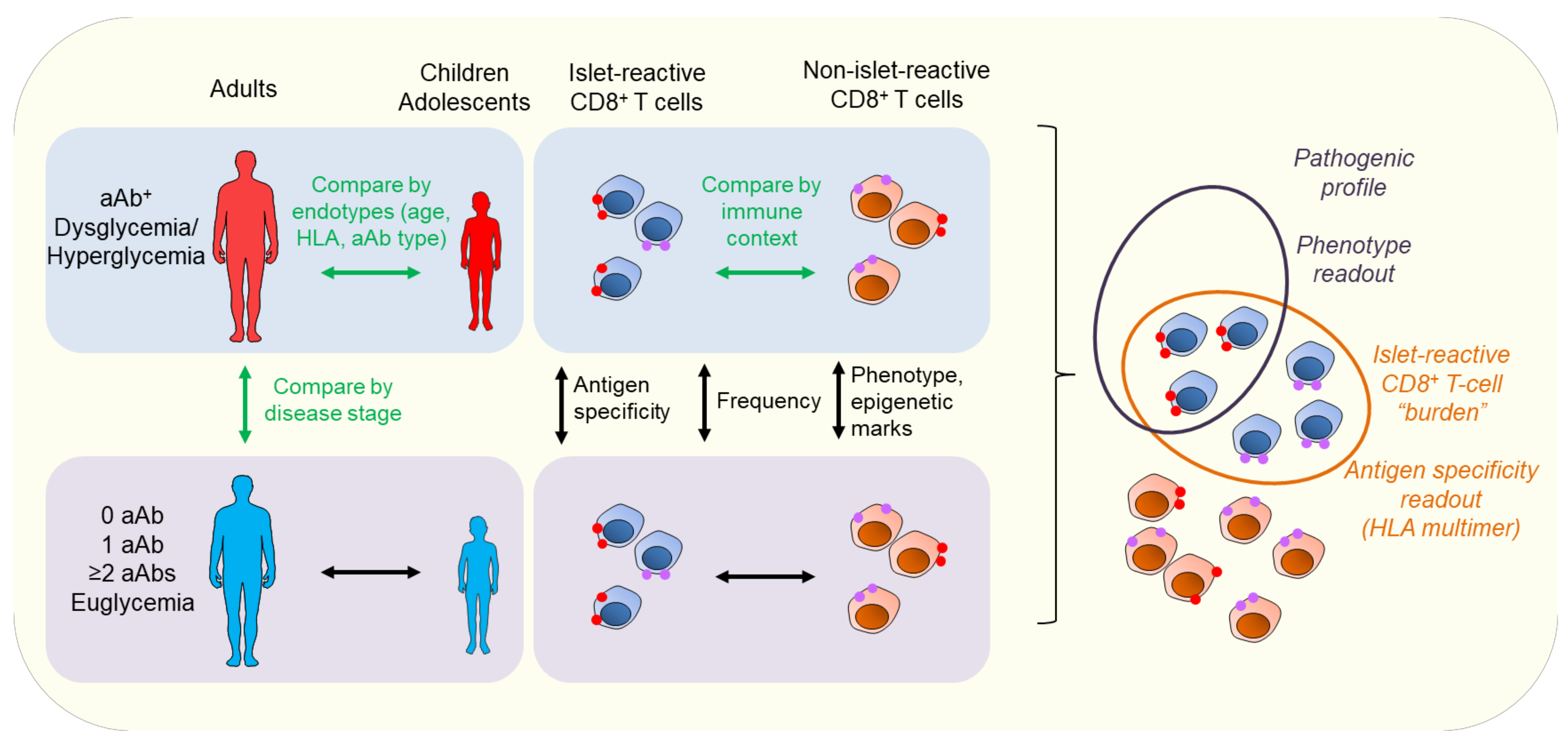
4. A Need for Biomarkers for Prognosis, Diagnosis, and Response to Therapy (“How”)
5. Towards Personalized Combination Therapy (“What”)
6. Conclusions and Perspectives
- (1)
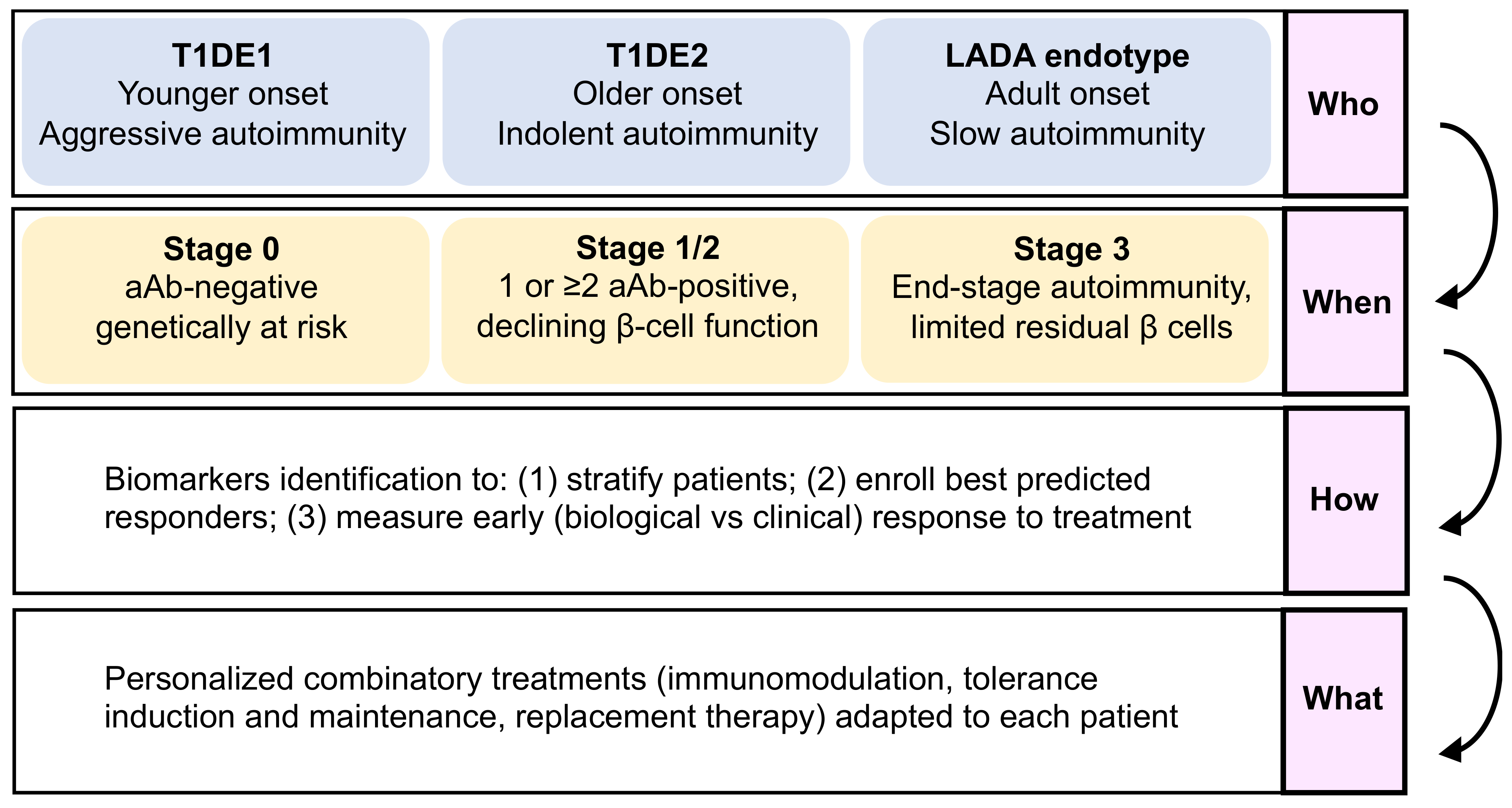
- The “who”, i.e., disease endotypes, which may help identify subgroups of patients with a younger age at diagnosis and a more aggressive autoimmune disease, who would benefit best from intense and multi-hit immunomodulation, versus patients with a more indolent pathology and a slower evolution.
- (2)
- The “when”, i.e., the optimal stage of intervention, as patients before aAb appearance (stage 0) are the best suited for immunotherapies aimed at blocking the development of autoimmune responses, with tolerogenic introduction of targeted antigens before T-cell priming and epitope spreading occurs, and with fully preserved β-cell function and mass. For patients with clinically overt T1D (stage 3), β-cell replacement combined with tolerance maintaining protocols may provide a better option.
- (3)
- The “how” should take advantage of omics data to identify biomarkers that will assist with stratifying patients and identifying responders and the best combination therapy to use (the “what”).
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carré, A.; Richardson, S.J.; Larger, E.; Mallone, R. Presumption of Guilt for T Cells in Type 1 Diabetes: Lead Culprits or Partners in Crime Depending on Age of Onset? Diabetologia 2021, 64, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Mallone, R.; Eizirik, D.L. Presumption of Innocence for Beta Cells: Why Are They Vulnerable Autoimmune Targets in Type 1 Diabetes? Diabetologia 2020, 63, 1999–2006. [Google Scholar] [CrossRef] [PubMed]
- Bottazzo, G.F.; Florin-Christensen, A.; Doniach, D. Islet-Cell Antibodies in Diabetes Mellitus with Autoimmune Polyendocrine Deficiencies. Lancet 1974, 2, 1279–1283. [Google Scholar] [CrossRef]
- Pugliese, A.; Zeller, M.; Fernandez, A.; Zalcberg, L.J.; Bartlett, R.J.; Ricordi, C.; Pietropaolo, M.; Eisenbarth, G.S.; Bennett, S.T.; Patel, D.D. The Insulin Gene Is Transcribed in the Human Thymus and Transcription Levels Correlated with Allelic Variation at the INS VNTR-IDDM2 Susceptibility Locus for Type 1 Diabetes. Nat. Genet. 1997, 15, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Bottini, N.; Musumeci, L.; Alonso, A.; Rahmouni, S.; Nika, K.; Rostamkhani, M.; MacMurray, J.; Meloni, G.F.; Lucarelli, P.; Pellecchia, M.; et al. A Functional Variant of Lymphoid Tyrosine Phosphatase Is Associated with Type I Diabetes. Nat. Genet. 2004, 36, 337–338. [Google Scholar] [CrossRef]
- Norris, J.M.; Johnson, R.K.; Stene, L.C. Type 1 Diabetes—Early Life Origins and Changing Epidemiology. Lancet Diabetes Endocrinol. 2020, 8, 226–238. [Google Scholar] [CrossRef]
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, Å.; et al. Staging Presymptomatic Type 1 Diabetes: A Scientific Statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974. [Google Scholar] [CrossRef] [Green Version]
- Lind, M.; Svensson, A.-M.; Kosiborod, M.; Gudbjörnsdottir, S.; Pivodic, A.; Wedel, H.; Dahlqvist, S.; Clements, M.; Rosengren, A. Glycemic Control and Excess Mortality in Type 1 Diabetes. N. Engl. J. Med. 2014, 371, 1972–1982. [Google Scholar] [CrossRef]
- Rawshani, A.; Sattar, N.; Franzén, S.; Rawshani, A.; Hattersley, A.T.; Svensson, A.-M.; Eliasson, B.; Gudbjörnsdottir, S. Excess Mortality and Cardiovascular Disease in Young Adults with Type 1 Diabetes in Relation to Age at Onset: A Nationwide, Register-Based Cohort Study. Lancet 2018, 392, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Huo, L.; Harding, J.L.; Peeters, A.; Shaw, J.E.; Magliano, D.J. Life Expectancy of Type 1 Diabetic Patients during 1997–2010: A National Australian Registry-Based Cohort Study. Diabetologia 2016, 59, 1177–1185. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, M.; Ahmed, S.; Anderson, M.S.; Atkinson, M.A.; Becker, D.; Bingley, P.J.; Bosi, E.; Brusko, T.M.; DiMeglio, L.A.; Evans-Molina, C.; et al. Introducing the Endotype Concept to Address the Challenge of Disease Heterogeneity in Type 1 Diabetes. Diabetes Care 2020, 43, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Orabona, C.; Mondanelli, G.; Puccetti, P.; Grohmann, U. Immune Checkpoint Molecules, Personalized Immunotherapy, and Autoimmune Diabetes. Trends Mol. Med. 2018, 24, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Hagopian, W.; Auger, J.A.; Poumian-Ruiz, E.; Taylor, L.; Donaldson, D.; Gitelman, S.E.; Harlan, D.M.; Xu, D.; Zivin, R.A.; et al. Anti-CD3 Monoclonal Antibody in New-Onset Type 1 Diabetes Mellitus. N. Engl. J. Med. 2002, 346, 1692–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keymeulen, B.; Vandemeulebroucke, E.; Ziegler, A.G.; Mathieu, C.; Kaufman, L.; Hale, G.; Gorus, F.; Goldman, M.; Walter, M.; Candon, S.; et al. Insulin Needs after CD3-Antibody Therapy in New-Onset Type 1 Diabetes. N. Engl. J. Med. 2005, 352, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Orban, T.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Marks, J.B.; Monzavi, R.; et al. Co-Stimulation Modulation with Abatacept in Patients with Recent-Onset Type 1 Diabetes: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet 2011, 378, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Rigby, M.R.; DiMeglio, L.A.; Rendell, M.S.; Felner, E.I.; Dostou, J.M.; Gitelman, S.E.; Patel, C.M.; Griffin, K.J.; Tsalikian, E.; Gottlieb, P.A.; et al. Targeting of Memory T Cells with Alefacept in New-Onset Type 1 Diabetes (T1DAL Study): 12 Month Results of a Randomised, Double-Blind, Placebo-Controlled Phase 2 Trial. Lancet Diabetes Endocrinol. 2013, 1, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Gitelman, S.E.; Gottlieb, P.A.; Rigby, M.R.; Felner, E.I.; Willi, S.M.; Fisher, L.K.; Moran, A.; Gottschalk, M.; Moore, W.V.; Pinckney, A.; et al. Antithymocyte Globulin Treatment for Patients with Recent-Onset Type 1 Diabetes: 12-Month Results of a Randomised, Placebo-Controlled, Phase 2 Trial. Lancet Diabetes Endocrinol. 2013, 1, 306–316. [Google Scholar] [CrossRef]
- Pescovitz, M.D.; Greenbaum, C.J.; Krause-Steinrauf, H.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; McGee, P.F.; Moran, A.M.; et al. Rituximab, B-Lymphocyte Depletion, and Preservation of Beta-Cell Function. N. Engl. J. Med. 2009, 361, 2143–2152. [Google Scholar] [CrossRef]
- Quattrin, T.; Haller, M.J.; Steck, A.K.; Felner, E.I.; Li, Y.; Xia, Y.; Leu, J.H.; Zoka, R.; Hedrick, J.A.; Rigby, M.R.; et al. Golimumab and Beta-Cell Function in Youth with New-Onset Type 1 Diabetes. N. Engl. J. Med. 2020, 383, 2007–2017. [Google Scholar] [CrossRef]
- Rapini, N.; Schiaffini, R.; Fierabracci, A. Immunotherapy Strategies for the Prevention and Treatment of Distinct Stages of Type 1 Diabetes: An Overview. Int. J. Mol. Sci. 2020, 21, 2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatenoud, L.; Primo, J.; Bach, J.F. CD3 Antibody-Induced Dominant Self Tolerance in Overtly Diabetic NOD Mice. J. Immunol. 1997, 158, 2947–2954. [Google Scholar]
- Bonifacio, E.; Ziegler, A.-G.; Klingensmith, G.; Schober, E.; Bingley, P.J.; Rottenkolber, M.; Theil, A.; Eugster, A.; Puff, R.; Peplow, C.; et al. Effects of High-Dose Oral Insulin on Immune Responses in Children at High Risk for Type 1 Diabetes: The Pre-POINT Randomized Clinical Trial. JAMA 2015, 313, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Roep, B.O.; Wheeler, D.C.S.; Peakman, M. Antigen-Based Immune Modulation Therapy for Type 1 Diabetes: The Era of Precision Medicine. Lancet Diabetes Endocrinol. 2019, 7, 65–74. [Google Scholar] [CrossRef]
- Ziegler, A.-G.; Bonifacio, E.; BABYDIAB-BABYDIET Study Group. Age-Related Islet Autoantibody Incidence in Offspring of Patients with Type 1 Diabetes. Diabetologia 2012, 55, 1937–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikka, V.; Näntö-Salonen, K.; Saarinen, M.; Simell, T.; Ilonen, J.; Hyöty, H.; Veijola, R.; Knip, M.; Simell, O. Early Seroconversion and Rapidly Increasing Autoantibody Concentrations Predict Prepubertal Manifestation of Type 1 Diabetes in Children at Genetic Risk. Diabetologia 2012, 55, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Assfalg, R.; Knoop, J.; Hoffman, K.L.; Pfirrmann, M.; Zapardiel-Gonzalo, J.M.; Hofelich, A.; Eugster, A.; Weigelt, M.; Matzke, C.; Reinhardt, J.; et al. Oral Insulin Immunotherapy in Children at Risk for Type 1 Diabetes in a Randomised Controlled Trial. Diabetologia 2021, 64, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Culina, S.; Gupta, N.; Boisgard, R.; Afonso, G.; Gagnerault, M.-C.; Dimitrov, J.; Østerbye, T.; Justesen, S.; Luce, S.; Attias, M.; et al. Materno-Fetal Transfer of Preproinsulin Through the Neonatal Fc Receptor Prevents Autoimmune Diabetes. Diabetes 2015, 64, 3532–3542. [Google Scholar] [CrossRef] [Green Version]
- Corcos, N.; Culina, S.; Deligne, C.; Lavaud, C.; You, S.; Mallone, R. Oral Fc-Coupled Preproinsulin Achieves Systemic and Thymic Delivery Through the Neonatal Fc Receptor and Partially Delays Autoimmune Diabetes. Front. Immunol. 2021, 12, 616215. [Google Scholar] [CrossRef]
- Ziegler, A.-G.; Kick, K.; Bonifacio, E.; Haupt, F.; Hippich, M.; Dunstheimer, D.; Lang, M.; Laub, O.; Warncke, K.; Lange, K.; et al. Yield of a Public Health Screening of Children for Islet Autoantibodies in Bavaria, Germany. JAMA 2020, 323, 339–351. [Google Scholar] [CrossRef]
- Cortez, F.D.J.; Gebhart, D.; Robinson, P.V.; Seftel, D.; Pourmandi, N.; Owyoung, J.; Bertozzi, C.R.; Wilson, D.M.; Maahs, D.M.; Buckingham, B.A.; et al. Sensitive Detection of Multiple Islet Autoantibodies in Type 1 Diabetes Using Small Sample Volumes by Agglutination-PCR. PLoS ONE 2020, 15, e0242049. [Google Scholar] [CrossRef] [PubMed]
- Kallionpää, H.; Elo, L.L.; Laajala, E.; Mykkänen, J.; Ricaño-Ponce, I.; Vaarma, M.; Laajala, T.D.; Hyöty, H.; Ilonen, J.; Veijola, R.; et al. Innate Immune Activity Is Detected Prior to Seroconversion in Children with HLA-Conferred Type 1 Diabetes Susceptibility. Diabetes 2014, 63, 2402–2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, R.C.; Guo, H.; Coulson, R.M.R.; Smyth, D.J.; Pekalski, M.L.; Burren, O.S.; Cutler, A.J.; Doecke, J.D.; Flint, S.; McKinney, E.F.; et al. A Type I Interferon Transcriptional Signature Precedes Autoimmunity in Children Genetically at Risk for Type 1 Diabetes. Diabetes 2014, 63, 2538–2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newby, B.N.; Mathews, C.E. Type I Interferon Is a Catastrophic Feature of the Diabetic Islet Microenvironment. Front. Endocrinol. 2017, 8, 232. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.J.; Rodrigues, K.B.; Quiel, J.A.; Liu, Y.; Bhargava, V.; Zhao, Y.; Hotta-Iwamura, C.; Shih, H.-Y.; Lau-Kilby, A.W.; Malloy, A.M.; et al. Restoration of the Type I IFN-IL-1 Balance through Targeted Blockade of PTGER4 Inhibits Autoimmunity in NOD Mice. JCI Insight 2018, 3, 97843. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Xu, B.; Michie, S.A.; Rubins, K.H.; Schreriber, R.D.; McDevitt, H.O. Interferon-Alpha Initiates Type 1 Diabetes in Nonobese Diabetic Mice. Proc. Natl. Acad. Sci. USA 2008, 105, 12439–12444. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, M.; Buckner, J.H.; Levings, M.K.; Richardson, S.J.; Wong, F.S.; Tree, T.I. Identifying the ‘Achilles Heel’ of Type 1 Diabetes. Clin. Exp. Immunol. 2021, 204, 167–178. [Google Scholar] [CrossRef]
- Leete, P.; Oram, R.A.; McDonald, T.J.; Shields, B.M.; Ziller, C.; TIGI Study Team; Hattersley, A.T.; Richardson, S.J.; Morgan, N.G. Studies of Insulin and Proinsulin in Pancreas and Serum Support the Existence of Aetiopathological Endotypes of Type 1 Diabetes Associated with Age at Diagnosis. Diabetologia 2020, 63, 1258–1267. [Google Scholar] [CrossRef] [Green Version]
- Hao, W.; Gitelman, S.; DiMeglio, L.A.; Boulware, D.; Greenbaum, C.J. Type 1 Diabetes TrialNet Study Group Fall in C-Peptide During First 4 Years from Diagnosis of Type 1 Diabetes: Variable Relation to Age, HbA1c, and Insulin Dose. Diabetes Care 2016, 39, 1664–1670. [Google Scholar] [CrossRef] [Green Version]
- Frohnert, B.I.; Ide, L.; Dong, F.; Barón, A.E.; Steck, A.K.; Norris, J.M.; Rewers, M.J. Late-Onset Islet Autoimmunity in Childhood: The Diabetes Autoimmunity Study in the Young (DAISY). Diabetologia 2017, 60, 998–1006. [Google Scholar] [CrossRef]
- Leete, P.; Mallone, R.; Richardson, S.J.; Sosenko, J.M.; Redondo, M.J.; Evans-Molina, C. The Effect of Age on the Progression and Severity of Type 1 Diabetes: Potential Effects on Disease Mechanisms. Curr. Diabetes Rep. 2018, 18, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leete, P.; Willcox, A.; Krogvold, L.; Dahl-Jørgensen, K.; Foulis, A.K.; Richardson, S.J.; Morgan, N.G. Differential Insulitic Profiles Determine the Extent of β-Cell Destruction and the Age at Onset of Type 1 Diabetes. Diabetes 2016, 65, 1362–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufort, M.J.; Greenbaum, C.J.; Speake, C.; Linsley, P.S. Cell Type-Specific Immune Phenotypes Predict Loss of Insulin Secretion in New-Onset Type 1 Diabetes. JCI Insight 2019, 4, 125556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, K.A.; Richardson, S.J.; Russell, M.A.; Morgan, N.G. Expression of CD47 in the Pancreatic β-Cells of People with Recent-Onset Type 1 Diabetes Varies According to Disease Endotype. Diabet. Med. J. Br. Diabet. Assoc. 2021, e14724. [Google Scholar] [CrossRef]
- Buzzetti, R.; Zampetti, S.; Maddaloni, E. Adult-Onset Autoimmune Diabetes: Current Knowledge and Implications for Management. Nat. Rev. Endocrinol. 2017, 13, 674–686. [Google Scholar] [CrossRef]
- Faucher, P.; Beuvon, F.; Fignani, D.; Sebastiani, G.; Afonso, G.; Zhou, Z.; Dousset, B.; Boitard, C.; Dotta, F.; Mallone, R.; et al. Immunoregulated Insulitis and Slow-Progressing Type 1 Diabetes after Duodenopancreatectomy. Diabetologia 2021, 64, 2731–2740. [Google Scholar] [CrossRef]
- McCarthy, M.I. Painting a New Picture of Personalised Medicine for Diabetes. Diabetologia 2017, 60, 793–799. [Google Scholar] [CrossRef] [Green Version]
- Arif, S.; Leete, P.; Nguyen, V.; Marks, K.; Nor, N.M.; Estorninho, M.; Kronenberg-Versteeg, D.; Bingley, P.J.; Todd, J.A.; Guy, C.; et al. Blood and Islet Phenotypes Indicate Immunological Heterogeneity in Type 1 Diabetes. Diabetes 2014, 63, 3835–3845. [Google Scholar] [CrossRef] [Green Version]
- Sherry, N.; Hagopian, W.; Ludvigsson, J.; Jain, S.M.; Wahlen, J.; Ferry, R.J.; Bode, B.; Aronoff, S.; Holland, C.; Carlin, D.; et al. Teplizumab for Treatment of Type 1 Diabetes (Protégé Study): 1-Year Results from a Randomised, Placebo-Controlled Trial. Lancet 2011, 378, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Linsley, P.S.; Greenbaum, C.J.; Speake, C.; Long, S.A.; Dufort, M.J. B Lymphocyte Alterations Accompany Abatacept Resistance in New-Onset Type 1 Diabetes. JCI Insight 2019, 4, 126136. [Google Scholar] [CrossRef] [Green Version]
- Vella, A.; Cooper, J.D.; Lowe, C.E.; Walker, N.; Nutland, S.; Widmer, B.; Jones, R.; Ring, S.M.; McArdle, W.; Pembrey, M.E.; et al. Localization of a Type 1 Diabetes Locus in the IL2RA/CD25 Region by Use of Tag Single-Nucleotide Polymorphisms. Am. J. Hum. Genet. 2005, 76, 773–779. [Google Scholar] [CrossRef] [Green Version]
- Qu, H.-Q.; Montpetit, A.; Ge, B.; Hudson, T.J.; Polychronakos, C. Toward Further Mapping of the Association between the IL2RA Locus and Type 1 Diabetes. Diabetes 2007, 56, 1174–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inshaw, J.R.J.; Cutler, A.J.; Crouch, D.J.M.; Wicker, L.S.; Todd, J.A. Genetic Variants Predisposing Most Strongly to Type 1 Diabetes Diagnosed Under Age 7 Years Lie Near Candidate Genes That Function in the Immune System and in Pancreatic β-Cells. Diabetes Care 2020, 43, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Rosenzwajg, M.; Churlaud, G.; Mallone, R.; Six, A.; Derian, N.; Chaara, W.; Lorenzon, R.; Long, A.; Buckner, J.; Afonso, G.; et al. Low-Dose Interleukin-2 Fosters a Dose-Dependent Regulatory T Cell Tuned Milieu in T1D Patients. J. Autoimmun. 2015, 58, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Hartemann, A.; Bensimon, G.; Payan, C.A.; Jacqueminet, S.; Bourron, O.; Nicolas, N.; Fonfrede, M.; Rosenzwajg, M.; Bernard, C.; Klatzmann, D. Low-Dose Interleukin 2 in Patients with Type 1 Diabetes: A Phase 1/2 Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Diabetes Endocrinol. 2013, 1, 295–305. [Google Scholar] [CrossRef]
- Rosenzwajg, M.; Salet, R.; Lorenzon, R.; Tchitchek, N.; Roux, A.; Bernard, C.; Carel, J.-C.; Storey, C.; Polak, M.; Beltrand, J.; et al. Low-Dose IL-2 in Children with Recently Diagnosed Type 1 Diabetes: A Phase I/II Randomised, Double-Blind, Placebo-Controlled, Dose-Finding Study. Diabetologia 2020, 63, 1808–1821. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Hiam-Galvez, K.J.; Mowery, C.T.; Herold, K.C.; Gitelman, S.E.; Esensten, J.H.; Liu, W.; Lares, A.P.; Leinbach, A.S.; Lee, M.; et al. The Effect of Low-Dose IL-2 and Treg Adoptive Cell Therapy in Patients with Type 1 Diabetes. JCI Insight 2021, 6, e147474. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Howson, J.M.M.; Esposito, L.; Heward, J.; Snook; Chamberlain, G.; Rainbow, D.B.; Hunter, K.M.D.; Smith, A.N.; Di Genova, G.; et al. Association of the T-Cell Regulatory Gene CTLA4 with Susceptibility to Autoimmune Disease. Nature 2003, 423, 506–511. [Google Scholar] [CrossRef]
- Nielsen, C.; Hansen, D.; Husby, S.; Jacobsen, B.B.; Lillevang, S.T. Association of a Putative Regulatory Polymorphism in the PD-1 Gene with Susceptibility to Type 1 Diabetes. Tissue Antigens 2003, 62, 492–497. [Google Scholar] [CrossRef]
- Gu, Y.; Xiao, L.; Gu, W.; Chen, S.; Feng, Y.; Wang, J.; Wang, Z.; Cai, Y.; Chen, H.; Xu, X.; et al. Rs2227982 and Rs2227981 in PDCD1 Gene Are Functional SNPs Associated with T1D Risk in East Asian. Acta Diabetol. 2018, 55, 813–819. [Google Scholar] [CrossRef]
- Falcone, M.; Fousteri, G. Role of the PD-1/PD-L1 Dyad in the Maintenance of Pancreatic Immune Tolerance for Prevention of Type 1 Diabetes. Front. Endocrinol. 2020, 11, 569. [Google Scholar] [CrossRef] [PubMed]
- Perdigoto, A.L.; Preston-Hurlburt, P.; Clark, P.; Long, S.A.; Linsley, P.S.; Harris, K.M.; Gitelman, S.E.; Greenbaum, C.J.; Gottlieb, P.A.; Hagopian, W.; et al. Treatment of Type 1 Diabetes with Teplizumab: Clinical and Immunological Follow-up after 7 Years from Diagnosis. Diabetologia 2019, 62, 655–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, S.A.; Thorpe, J.; Herold, K.C.; Ehlers, M.; Sanda, S.; Lim, N.; Linsley, P.S.; Nepom, G.T.; Harris, K.M. Remodeling T Cell Compartments during Anti-CD3 Immunotherapy of Type 1 Diabetes. Cell. Immunol. 2017, 319, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Orabona, C.; Mondanelli, G.; Pallotta, M.T.; Carvalho, A.; Albini, E.; Fallarino, F.; Vacca, C.; Volpi, C.; Belladonna, M.L.; Berioli, M.G.; et al. Deficiency of Immunoregulatory Indoleamine 2,3-Dioxygenase 1 in Juvenile Diabetes. JCI Insight 2021, 3, e96244. [Google Scholar] [CrossRef]
- Hannelius, U.; Beam, C.A.; Ludvigsson, J. Efficacy of GAD-Alum Immunotherapy Associated with HLA-DR3-DQ2 in Recently Diagnosed Type 1 Diabetes. Diabetologia 2020, 63, 2177–2181. [Google Scholar] [CrossRef]
- Noble, J.A.; Valdes, A.M. Genetics of the HLA Region in the Prediction of Type 1 Diabetes. Curr. Diabetes Rep. 2011, 11, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, R.C.; Lanzoni, G.; Russell, M.A.; Gerling, I.; Richardson, S.J. What the HLA-I!-Classical and Non-Classical HLA Class I and Their Potential Roles in Type 1 Diabetes. Curr. Diabetes Rep. 2019, 19, 159. [Google Scholar] [CrossRef] [Green Version]
- Ludvigsson, J.; Wahlberg, J.; Casas, R. Intralymphatic Injection of Autoantigen in Type 1 Diabetes. N. Engl. J. Med. 2017, 376, 697–699. [Google Scholar] [CrossRef] [Green Version]
- Culina, S.; Mallone, R. Immune Biomarkers in Immunotherapeutic Trials for Type 1 Diabetes: Cui Prodest? Diabetes Metab. 2012, 38, 379–385. [Google Scholar] [CrossRef]
- Gonzalez-Duque, S.; Azoury, M.E.; Colli, M.L.; Afonso, G.; Turatsinze, J.-V.; Nigi, L.; Lalanne, A.I.; Sebastiani, G.; Carré, A.; Pinto, S.; et al. Conventional and Neo-Antigenic Peptides Presented by β Cells Are Targeted by Circulating Naïve CD8+ T Cells in Type 1 Diabetic and Healthy Donors. Cell Metab. 2018, 28, 946–960.e6. [Google Scholar] [CrossRef] [Green Version]
- Azoury, M.E.; Samassa, F.; Buitinga, M.; Nigi, L.; Brusco, N.; Callebaut, A.; Giraud, M.; Irla, M.; Lalanne, A.I.; Carré, A.; et al. CD8+ T Cells Variably Recognize Native Versus Citrullinated GRP78 Epitopes in Type 1 Diabetes. Diabetes 2021, 70, 2879–2891. [Google Scholar] [CrossRef] [PubMed]
- James, E.A.; Mallone, R.; Kent, S.C.; DiLorenzo, T.P. T-Cell Epitopes and Neo-Epitopes in Type 1 Diabetes: A Comprehensive Update and Reappraisal. Diabetes 2020, 69, 1311–1335. [Google Scholar] [CrossRef] [PubMed]
- Azoury, M.E.; Tarayrah, M.; Afonso, G.; Pais, A.; Colli, M.L.; Maillard, C.; Lavaud, C.; Alexandre-Heymann, L.; Gonzalez-Duque, S.; Verdier, Y.; et al. Peptides Derived from Insulin Granule Proteins Are Targeted by CD8+ T Cells Across MHC Class I Restrictions in Humans and NOD Mice. Diabetes 2020, 69, 2678–2690. [Google Scholar] [CrossRef] [PubMed]
- Culina, S.; Lalanne, A.I.; Afonso, G.; Cerosaletti, K.; Pinto, S.; Sebastiani, G.; Kuranda, K.; Nigi, L.; Eugster, A.; Østerbye, T.; et al. Islet-Reactive CD8 + T Cell Frequencies in the Pancreas, but Not in Blood, Distinguish Type 1 Diabetic Patients from Healthy Donors. Sci. Immunol. 2018, 20, eaao4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Broek, T.; Borghans, J.A.M.; van Wijk, F. The Full Spectrum of Human Naive T Cells. Nat. Rev. Immunol. 2018, 18, 363–373. [Google Scholar] [CrossRef]
- Vignali, D.; Cantarelli, E.; Bordignon, C.; Canu, A.; Citro, A.; Annoni, A.; Piemonti, L.; Monti, P. Detection and Characterization of CD8+ Autoreactive Memory Stem T Cells in Patients with Type 1 Diabetes. Diabetes 2018, 67, 936–945. [Google Scholar] [CrossRef] [Green Version]
- Gearty, S.V.; Dündar, F.; Zumbo, P.; Espinosa-Carrasco, G.; Shakiba, M.; Sanchez-Rivera, F.J.; Socci, N.D.; Trivedi, P.; Lowe, S.W.; Lauer, P.; et al. An Autoimmune Stem-like CD8 T Cell Population Drives Type 1 Diabetes. Nature 2021, 602, 156–161. [Google Scholar] [CrossRef]
- Abdelsamed, H.A.; Zebley, C.C.; Nguyen, H.; Rutishauser, R.L.; Fan, Y.; Ghoneim, H.E.; Crawford, J.C.; Alfei, F.; Alli, S.; Ribeiro, S.P.; et al. Beta Cell-Specific CD8+ T Cells Maintain Stem-Cell Memory-Associated Epigenetic Programs during Type 1 Diabetes. Nat. Immunol. 2020, 21, 578–587. [Google Scholar] [CrossRef]
- Huang, J.; Xiao, Y.; Xu, A.; Zhou, Z. Neutrophils in Type 1 Diabetes. J. Diabetes Investig. 2016, 7, 652–663. [Google Scholar] [CrossRef]
- Valle, A.; Giamporcaro, G.M.; Scavini, M.; Stabilini, A.; Grogan, P.; Bianconi, E.; Sebastiani, G.; Masini, M.; Maugeri, N.; Porretti, L.; et al. Reduction of Circulating Neutrophils Precedes and Accompanies Type 1 Diabetes. Diabetes 2013, 62, 2072–2077. [Google Scholar] [CrossRef] [Green Version]
- Nichols, B.E.; Hook, J.S.; Weng, K.; Ahn, C.; Moreland, J.G. Novel Neutrophil Phenotypic Signature in Pediatric Patients with Type 1 Diabetes and Diabetic Ketoacidosis. J. Leukoc. Biol. 2021, 111, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Popp, S.K.; Vecchio, F.; Brown, D.J.; Fukuda, R.; Suzuki, Y.; Takeda, Y.; Wakamatsu, R.; Sarma, M.A.; Garrett, J.; Giovenzana, A.; et al. Circulating Platelet-Neutrophil Aggregates Characterize the Development of Type 1 Diabetes in Humans and NOD Mice. JCI Insight 2022, 7, e153993. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, F.; Buono, N.L.; Stabilini, A.; Nigi, L.; Dufort, M.J.; Geyer, S.; Rancoita, P.M.; Cugnata, F.; Mandelli, A.; Valle, A.; et al. Abnormal Neutrophil Signature in the Blood and Pancreas of Presymptomatic and Symptomatic Type 1 Diabetes. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purohit, S.; Sharma, A.; Hopkins, D.; Steed, L.; Bode, B.; Anderson, S.W.; Reed, J.C.; Steed, R.D.; Yang, T.; She, J.-X. Large-Scale Discovery and Validation Studies Demonstrate Significant Reductions in Circulating Levels of IL8, IL-1Ra, MCP-1, and MIP-1β in Patients with Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2015, 100, E1179–E1187. [Google Scholar] [CrossRef] [Green Version]
- Yeung, W.-C.G.; Al-Shabeeb, A.; Pang, C.N.I.; Wilkins, M.R.; Catteau, J.; Howard, N.J.; Rawlinson, W.D.; Craig, M.E. Children with Islet Autoimmunity and Enterovirus Infection Demonstrate a Distinct Cytokine Profile. Diabetes 2012, 61, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Speake, C.; Ylescupidez, A.; Neiman, D.; Shemer, R.; Glaser, B.; Tersey, S.A.; Usmani-Brown, S.; Clark, P.; Wilhelm, J.J.; Bellin, M.D.; et al. Circulating Unmethylated Insulin DNA As a Biomarker of Human Beta Cell Death: A Multi-Laboratory Assay Comparison. J. Clin. Endocrinol. Metab. 2020, 105, 781–791. [Google Scholar] [CrossRef]
- Sims, E.K.; Chaudhry, Z.; Watkins, R.; Syed, F.; Blum, J.; Ouyang, F.; Perkins, S.M.; Mirmira, R.G.; Sosenko, J.; DiMeglio, L.A.; et al. Elevations in the Fasting Serum Proinsulin–to–C-Peptide Ratio Precede the Onset of Type 1 Diabetes. Diabetes Care 2016, 39, 1519–1526. [Google Scholar] [CrossRef] [Green Version]
- Sims, E.K.; Bahnson, H.T.; Nyalwidhe, J.; Haataja, L.; Davis, A.K.; Speake, C.; DiMeglio, L.A.; Blum, J.; Morris, M.A.; Mirmira, R.G.; et al. Proinsulin Secretion Is a Persistent Feature of Type 1 Diabetes. Diabetes Care 2018, 42, 258–264. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Calvo, T.; Chen, Y.-C.; Verchere, C.B.; Haataja, L.; Arvan, P.; Leete, P.; Richardson, S.J.; Morgan, N.G.; Qian, W.-J.; Pugliese, A.; et al. Altered β-Cell Prohormone Processing and Secretion in Type 1 Diabetes. Diabetes 2021, 70, 1038–1050. [Google Scholar] [CrossRef]
- Carré, A.; Mallone, R. Making Insulin and Staying Out of Autoimmune Trouble: The Beta-Cell Conundrum. Front. Immunol. 2021, 12, 996. [Google Scholar] [CrossRef]
- Crèvecoeur, I.; Rondas, D.; Mathieu, C.; Overbergh, L. The Beta-Cell in Type 1 Diabetes: What Have We Learned from Proteomic Studies? Proteom. Clin. Appl. 2015, 9, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Crèvecoeur, I.; Vig, S.; Mathieu, C.; Overbergh, L. Understanding Type 1 Diabetes through Proteomics. Expert Rev. Proteom. 2017, 14, 571–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, S.A.; Rich, S.S.; Wood, A.R.; Jones, S.E.; Beaumont, R.N.; Harrison, J.W.; Schneider, D.A.; Locke, J.M.; Tyrrell, J.; Weedon, M.N.; et al. Development and Standardization of an Improved Type 1 Diabetes Genetic Risk Score for Use in Newborn Screening and Incident Diagnosis. Diabetes Care 2019, 42, 200–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonifacio, E.; Beyerlein, A.; Hippich, M.; Winkler, C.; Vehik, K.; Weedon, M.N.; Laimighofer, M.; Hattersley, A.T.; Krumsiek, J.; Frohnert, B.I.; et al. Genetic Scores to Stratify Risk of Developing Multiple Islet Autoantibodies and Type 1 Diabetes: A Prospective Study in Children. PLoS Med. 2018, 15, e1002548. [Google Scholar] [CrossRef] [PubMed]
- Ferrat, L.A.; Vehik, K.; Sharp, S.A.; Lernmark, Å.; Rewers, M.J.; She, J.-X.; Ziegler, A.-G.; Toppari, J.; Akolkar, B.; Krischer, J.P.; et al. A Combined Risk Score Enhances Prediction of Type 1 Diabetes among Susceptible Children. Nat. Med. 2020, 26, 1247–1255. [Google Scholar] [CrossRef]
- Alcazar, O.; Hernandez, L.F.; Nakayasu, E.S.; Nicora, C.D.; Ansong, C.; Muehlbauer, M.J.; Bain, J.R.; Myer, C.J.; Bhattacharya, S.K.; Buchwald, P.; et al. Parallel Multi-Omics in High-Risk Subjects for the Identification of Integrated Biomarker Signatures of Type 1 Diabetes. Biomolecules 2021, 11, 383. [Google Scholar] [CrossRef]
- Edner, N.M.; Heuts, F.; Thomas, N.; Wang, C.J.; Petersone, L.; Kenefeck, R.; Kogimtzis, A.; Ovcinnikovs, V.; Ross, E.M.; Ntavli, E.; et al. Follicular Helper T Cell Profiles Predict Response to Costimulation Blockade in Type 1 Diabetes. Nat. Immunol. 2020, 21, 1244–1255. [Google Scholar] [CrossRef]
- Cabrera, S.M.; Engle, S.; Kaldunski, M.; Jia, S.; Geoffrey, R.; Simpson, P.; Szabo, A.; Speake, C.; Greenbaum, C.J.; Type 1 Diabetes TrialNet CTLA4-Ig (Abatacept) Study Group; et al. Innate Immune Activity as a Predictor of Persistent Insulin Secretion and Association with Responsiveness to CTLA4-Ig Treatment in Recent-Onset Type 1 Diabetes. Diabetologia 2018, 61, 2356–2370. [Google Scholar] [CrossRef] [Green Version]
- Diggins, K.E.; Serti, E.; Muir, V.; Rosasco, M.; Lu, T.; Balmas, E.; Nepom, G.; Long, S.A.; Linsley, P.S. Exhausted-like CD8+ T Cell Phenotypes Linked to C-Peptide Preservation in Alefacept-Treated T1D Subjects. JCI Insight 2021, 6, 142680. [Google Scholar] [CrossRef]
- Long, S.A.; Thorpe, J.; DeBerg, H.A.; Gersuk, V.; Eddy, J.; Harris, K.M.; Ehlers, M.; Herold, K.C.; Nepom, G.T.; Linsley, P.S. Partial Exhaustion of CD8 T Cells and Clinical Response to Teplizumab in New-Onset Type 1 Diabetes. Sci. Immunol. 2016, 1, eaai7793. [Google Scholar] [CrossRef] [Green Version]
- Sims, E.K.; Bundy, B.N.; Stier, K.; Serti, E.; Lim, N.; Long, S.A.; Geyer, S.M.; Moran, A.; Greenbaum, C.J.; Evans-Molina, C.; et al. Teplizumab Improves and Stabilizes Beta Cell Function in Antibody-Positive High-Risk Individuals. Sci. Transl. Med. 2021, 13, eabc8980. [Google Scholar] [CrossRef] [PubMed]
- Habib, T.; Long, S.A.; Samuels, P.L.; Brahmandam, A.; Tatum, M.; Funk, A.; Hocking, A.M.; Cerosaletti, K.; Mason, M.T.; Whalen, E.; et al. Dynamic Immune Phenotypes of B and T Helper Cells Mark Distinct Stages of T1D Progression. Diabetes 2019, 68, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Greenbaum, C.J.; Rosasco, M.; Presnell, S.; Herold, K.C.; Dufort, M.J. Elevated T Cell Levels in Peripheral Blood Predict Poor Clinical Response Following Rituximab Treatment in New-Onset Type 1 Diabetes. Genes Immun. 2019, 20, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Besançon, A.; Goncalves, T.; Valette, F.; Mary, C.; Vanhove, B.; Chatenoud, L.; You, S. A Selective CD28 Antagonist and Rapamycin Synergise to Protect against Spontaneous Autoimmune Diabetes in NOD Mice. Diabetologia 2018, 61, 1811–1816. [Google Scholar] [CrossRef] [Green Version]
- Manirarora, J.N.; Wei, C.-H. Combination Therapy Using IL-2/IL-2 Monoclonal Antibody Complexes, Rapamycin, and Islet Autoantigen Peptides Increases Regulatory T Cell Frequency and Protects against Spontaneous and Induced Type 1 Diabetes in Nonobese Diabetic Mice. J. Immunol. 2015, 195, 5203–5214. [Google Scholar] [CrossRef] [Green Version]
- Long, S.A.; Rieck, M.; Sanda, S.; Bollyky, J.B.; Samuels, P.L.; Goland, R.; Ahmann, A.; Rabinovitch, A.; Aggarwal, S.; Phippard, D.; et al. Rapamycin/IL-2 Combination Therapy in Patients with Type 1 Diabetes Augments Tregs yet Transiently Impairs β-Cell Function. Diabetes 2012, 61, 2340–2348. [Google Scholar] [CrossRef] [Green Version]
- Haller, M.J.; Long, S.A.; Blanchfield, J.L.; Schatz, D.A.; Skyler, J.S.; Krischer, J.P.; Bundy, B.N.; Geyer, S.M.; Warnock, M.V.; Miller, J.L.; et al. Low-Dose Anti-Thymocyte Globulin Preserves C-Peptide, Reduces HbA1c, and Increases Regulatory to Conventional T-Cell Ratios in New-Onset Type 1 Diabetes: Two-Year Clinical Trial Data. Diabetes 2019, 68, 1267–1276. [Google Scholar] [CrossRef] [Green Version]
- Haller, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Michels, A.W.; Rosenthal, S.M.; Shuster, J.J.; Zou, B.; Brusko, T.M.; Hulme, M.A.; Wasserfall, C.H.; et al. Anti-Thymocyte Globulin/G-CSF Treatment Preserves β Cell Function in Patients with Established Type 1 Diabetes. J. Clin. Investig. 2015, 125, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Matthews, J.B.; Staeva, T.P.; Bernstein, P.L.; Peakman, M.; von Herrath, M. ITN-JDRF Type 1 Diabetes Combination Therapy Assessment Group Developing Combination Immunotherapies for Type 1 Diabetes: Recommendations from the ITN-JDRF Type 1 Diabetes Combination Therapy Assessment Group. Clin. Exp. Immunol. 2010, 160, 176–184. [Google Scholar] [CrossRef]
- De Groot, P.; Nikolic, T.; Pellegrini, S.; Sordi, V.; Imangaliyev, S.; Rampanelli, E.; Hanssen, N.; Attaye, I.; Bakker, G.; Duinkerken, G.; et al. Faecal Microbiota Transplantation Halts Progression of Human New-Onset Type 1 Diabetes in a Randomised Controlled Trial. Gut 2021, 70, 92–105. [Google Scholar] [CrossRef]
- Ho, J.; Nicolucci, A.C.; Virtanen, H.; Schick, A.; Meddings, J.; Reimer, R.A.; Huang, C. Effect of Prebiotic on Microbiota, Intestinal Permeability, and Glycemic Control in Children with Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2019, 104, 4427–4440. [Google Scholar] [CrossRef] [PubMed]
- De Groot, P.F.; Nikolic, T.; Imangaliyev, S.; Bekkering, S.; Duinkerken, G.; Keij, F.M.; Herrema, H.; Winkelmeijer, M.; Kroon, J.; Levin, E.; et al. Oral Butyrate Does Not Affect Innate Immunity and Islet Autoimmunity in Individuals with Longstanding Type 1 Diabetes: A Randomised Controlled Trial. Diabetologia 2020, 63, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 Diabetes Immunotherapy Using Polyclonal Regulatory T Cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabello-Kindelan, C.; Mackey, S.; Sands, A.; Rodriguez, J.; Vazquez, C.; Pugliese, A.; Bayer, A.L. Immunomodulation Followed by Antigen-Specific Treg Infusion Controls Islet Autoimmunity. Diabetes 2020, 69, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Von Herrath, M.; Bain, S.C.; Bode, B.; Clausen, J.O.; Coppieters, K.; Gaysina, L.; Gumprecht, J.; Hansen, T.K.; Mathieu, C.; Morales, C.; et al. Anti-Interleukin-21 Antibody and Liraglutide for the Preservation of β-Cell Function in Adults with Recent-Onset Type 1 Diabetes: A Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet Diabetes Endocrinol. 2021, 9, 212–224. [Google Scholar] [CrossRef]
- Vantyghem, M.-C.; de Koning, E.J.P.; Pattou, F.; Rickels, M.R. Advances in β-Cell Replacement Therapy for the Treatment of Type 1 Diabetes. Lancet 2019, 394, 1274–1285. [Google Scholar] [CrossRef]
- Pellegrini, S.; Piemonti, L.; Sordi, V. Pluripotent Stem Cell Replacement Approaches to Treat Type 1 Diabetes. Curr. Opin. Pharmacol. 2018, 43, 20–26. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Biomarker | Drug | Patients Stage | Associated Phenotype | Reference |
|---|---|---|---|---|
| ICOS and PD-1 expression on T follicular helper | abatacept | New-onset T1D | Predict decreased response to treatment | [97] |
| B-cell-related genes | abatacept | New-onset T1D | Increased in non-responders | [50] |
| Neutrophil-related genes | abatacept | New-onset T1D | Increased in responders | [50] |
| TIGIT and KLRG1 on CD8+ T cells | alefacept | New-onset T1D | Increased in responders | [99] |
| TIGIT and KLRG1 on CD8+ T cells | teplizumab | New-onset T1D | Increased in responders | [100,101] |
| Low innate inflammatory signature | abatacept | New-onset T1D | Greater responders | [98] |
| HLADR4+ HLA-DR3− | teplizumab | At risk, ≥2 aAbs | Increased in responders | [15] |
| ZnT8 autoantibodies | teplizumab | At risk, ≥2 aAbs | Decreased in responders | [15] |
| PD-1 expression on CD4+ and CD8+ T cells | teplizumab | New-onset T1D (8 weeks) | Increased in responders | [62,63] |
| BCR signaling | rituximab | Established T1D | Increased in responders | [102] |
| Heterogenous T-cell signature | rituximab | New-onset T1D | Decreased efficacy | [103] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deligne, C.; You, S.; Mallone, R. Personalized Immunotherapies for Type 1 Diabetes: Who, What, When, and How? J. Pers. Med. 2022, 12, 542. https://doi.org/10.3390/jpm12040542
Deligne C, You S, Mallone R. Personalized Immunotherapies for Type 1 Diabetes: Who, What, When, and How? Journal of Personalized Medicine. 2022; 12(4):542. https://doi.org/10.3390/jpm12040542
Chicago/Turabian StyleDeligne, Claire, Sylvaine You, and Roberto Mallone. 2022. "Personalized Immunotherapies for Type 1 Diabetes: Who, What, When, and How?" Journal of Personalized Medicine 12, no. 4: 542. https://doi.org/10.3390/jpm12040542
APA StyleDeligne, C., You, S., & Mallone, R. (2022). Personalized Immunotherapies for Type 1 Diabetes: Who, What, When, and How? Journal of Personalized Medicine, 12(4), 542. https://doi.org/10.3390/jpm12040542