
Transthyretin Amyloid Cardiomyopathy: Impact of Transthyretin Amyloid Deposition in Myocardium on Cardiac Morphology and Function

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Population
2.2. Clinical Assessment
2.3. Histological Assessment
2.4. Immunohistochemical Analysis
2.5. Statistical Analysis
3. Results
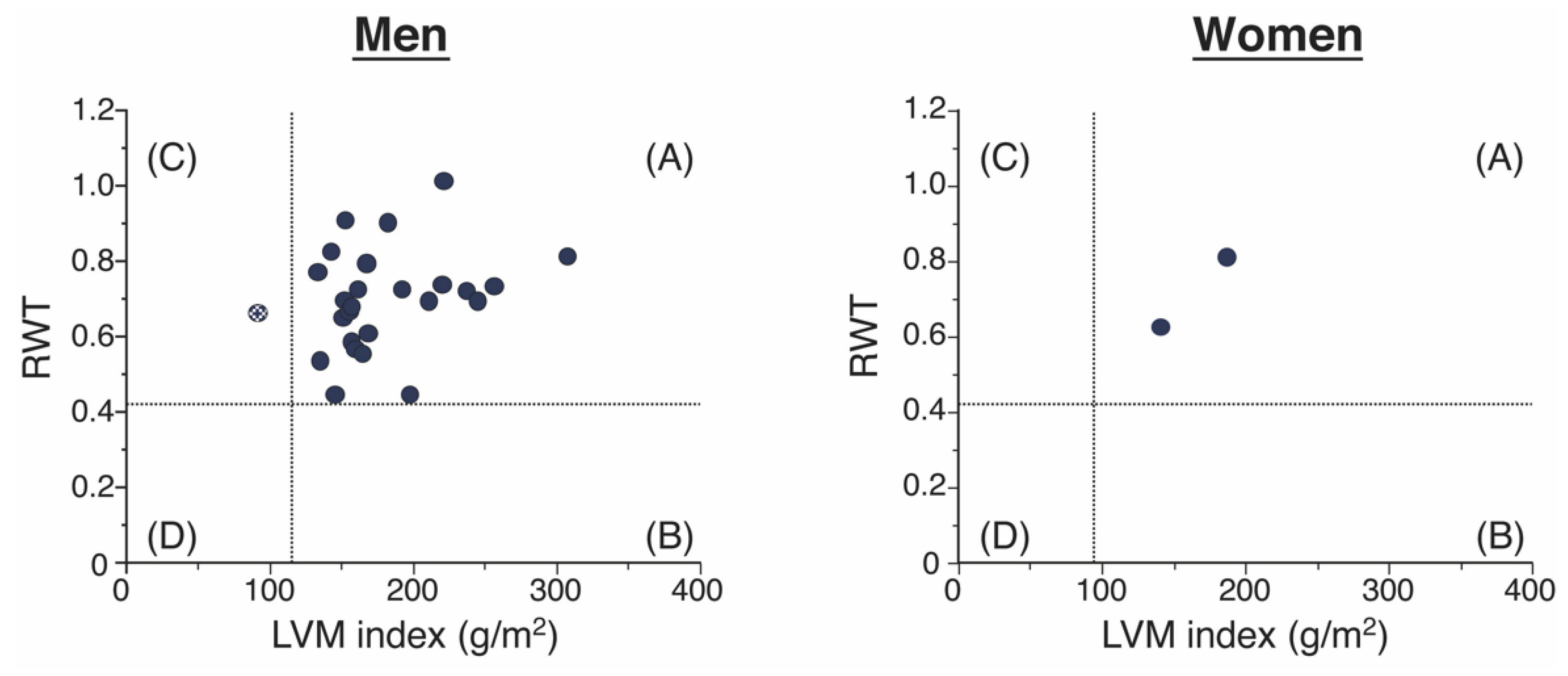
3.1. Clinical Characteristics and LV Geometries of the Patients with ATTR-CM
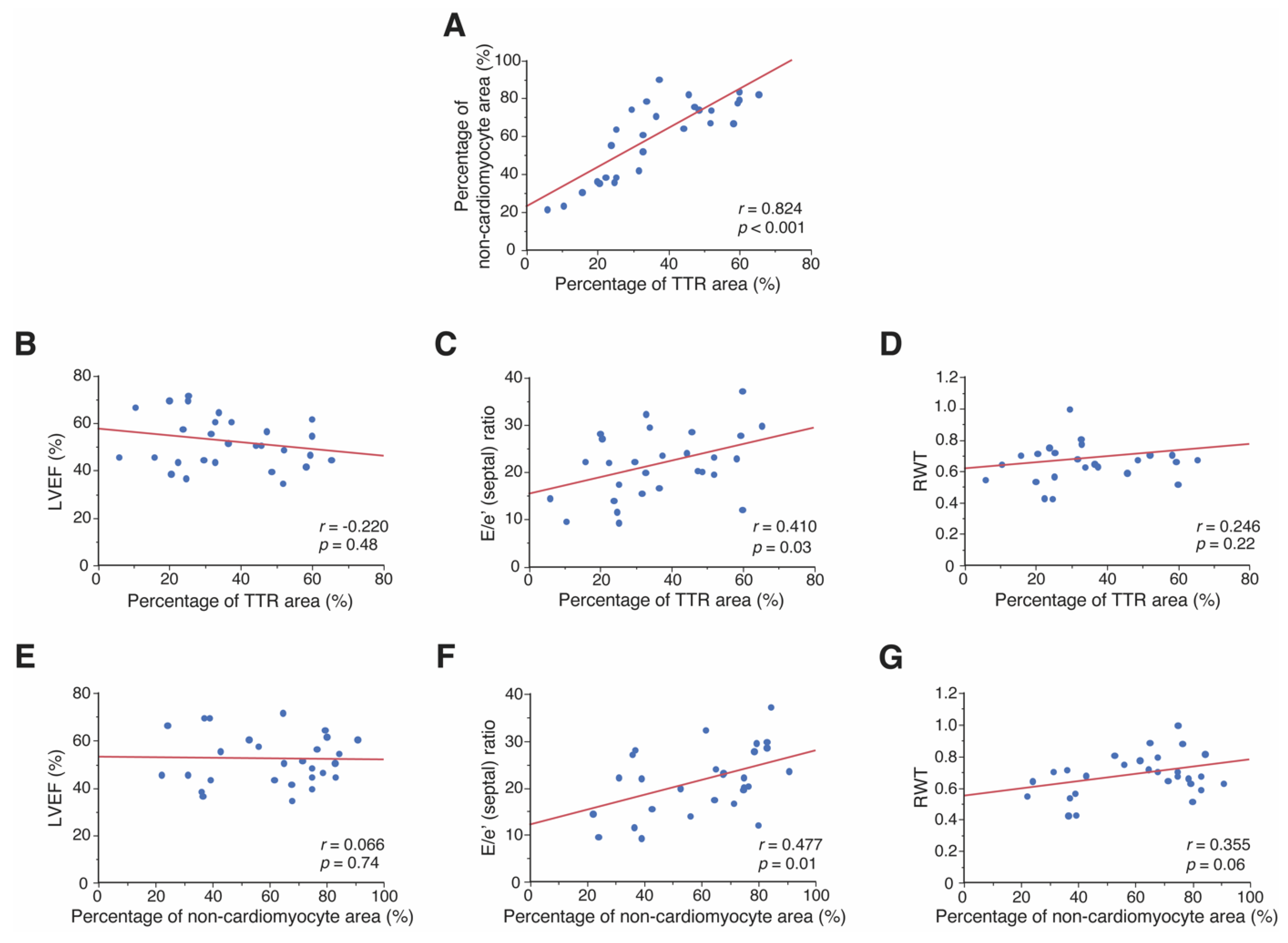
3.2. Relationship between Pathological Features and Cardiac Morphology and Function
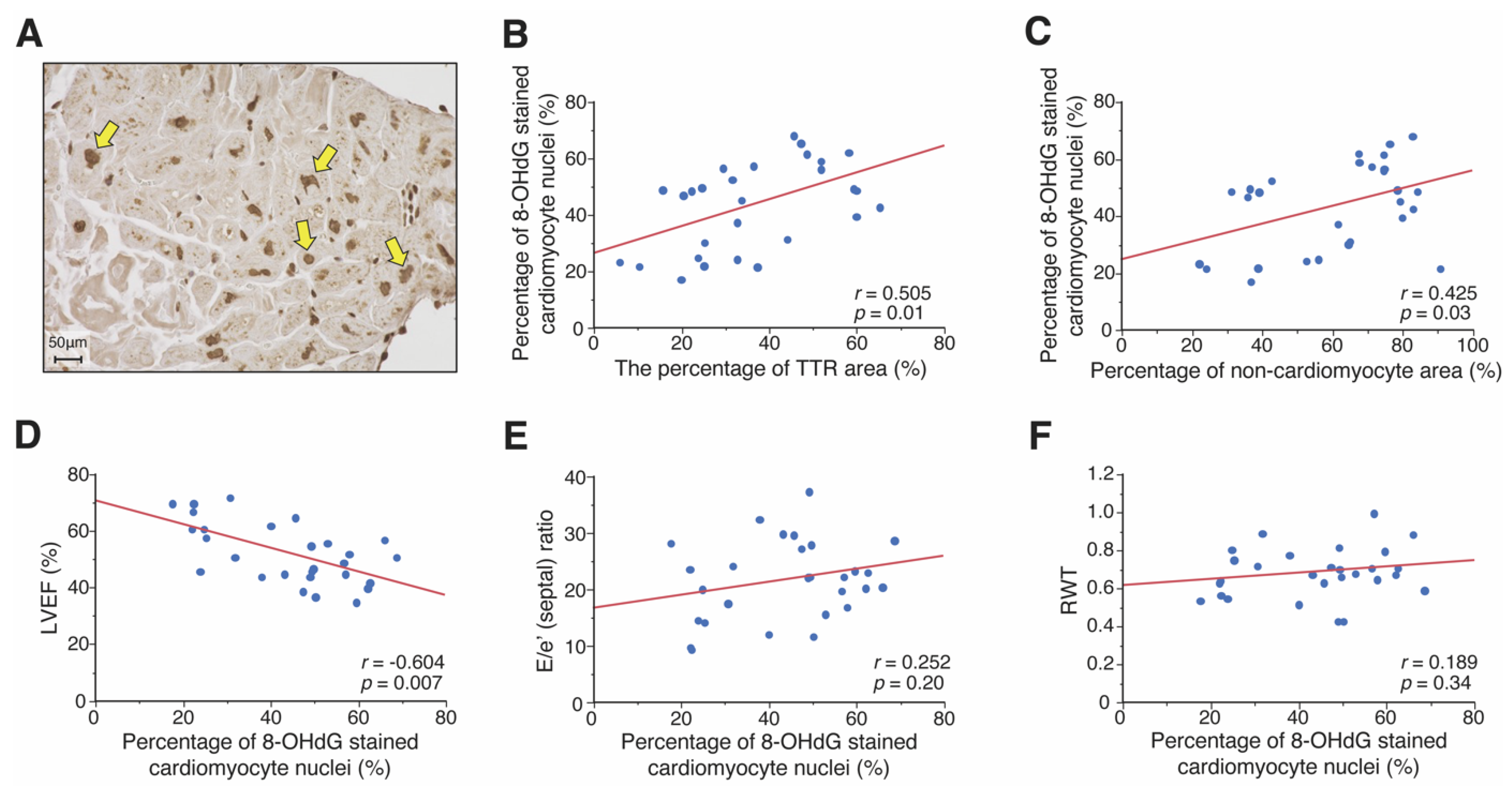
3.3. Association between ROS Accumulation and Pathological and Clinical Features
4. Discussion
4.1. Cardiac Morphology
4.2. Relationship between the Pathological Features and Cardiac Dysfunction
4.3. The Involvement of ROS Accumulation and Cardiac Dysfunction
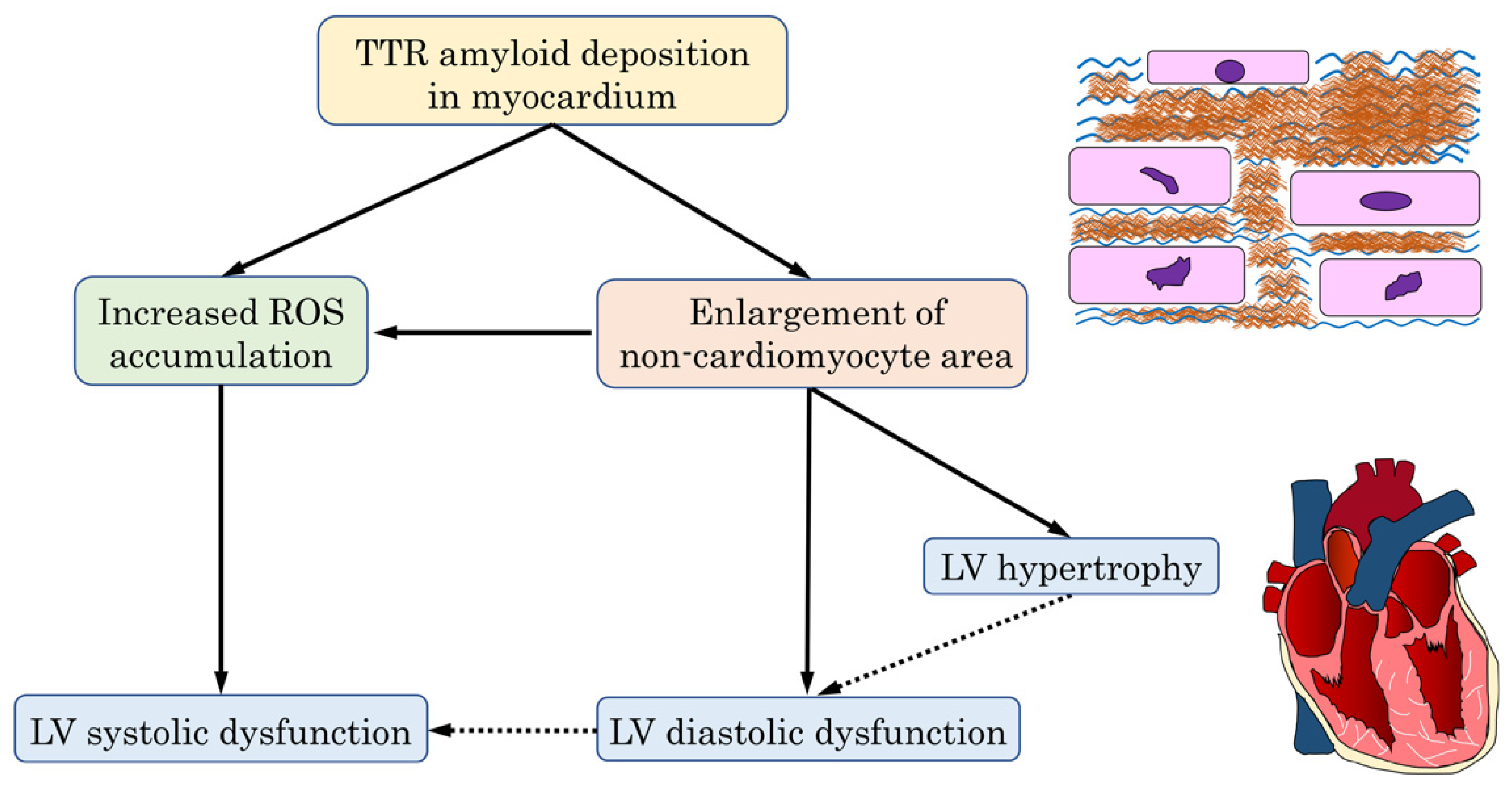
4.4. The Mechanism of Cardiac Dysfunction in the ATTR-CM Heart
4.5. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Merlini, G.; Bellotti, V. Molecular mechanisms of amyloidosis. N. Engl. J. Med. 2003, 349, 583–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oerlemans, M.; Rutten, K.H.G.; Minnema, M.C.; Raymakers, R.A.P.; Asselbergs, F.W.; de Jonge, N. Cardiac amyloidosis: The need for early diagnosis. Neth. Heart J. 2019, 27, 525–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqi, O.K.; Ruberg, F.L. Cardiac amyloidosis: An update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc. Med. 2018, 28, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Elliott, P.; Comenzo, R.; Semigran, M.; Rapezzi, C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation 2017, 135, 1357–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emdin, M.; Aimo, A.; Rapezzi, C.; Fontana, M.; Perfetto, F.; Seferović, P.M.; Barison, A.; Castiglione, V.; Vergaro, G.; Giannoni, A.; et al. Treatment of cardiac transthyretin amyloidosis: An update. Eur. Heart J. 2019, 40, 3699–3706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2872–2891. [Google Scholar] [CrossRef] [PubMed]
- Dubrey, S.W.; Hawkins, P.N.; Falk, R.H. Amyloid diseases of the heart: Assessment, diagnosis, and referral. Heart 2011, 97, 75–84. [Google Scholar] [CrossRef]
- Griffin, J.M.; Rosenthal, J.L.; Grodin, J.L.; Maurer, M.S.; Grogan, M.; Cheng, R.K. ATTR Amyloidosis: Current and Emerging Management Strategies: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2021, 3, 488–505. [Google Scholar] [CrossRef]
- González-López, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur. Heart J. 2015, 36, 2585–2594. [Google Scholar] [CrossRef] [Green Version]
- Treibel, T.A.; Fontana, M.; Gilbertson, J.A.; Castelletti, S.; White, S.K.; Scully, P.R.; Roberts, N.; Hutt, D.F.; Rowczenio, D.M.; Whelan, C.J.; et al. Occult Transthyretin Cardiac Amyloid in Severe Calcific Aortic Stenosis: Prevalence and Prognosis in Patients Undergoing Surgical Aortic Valve Replacement. Circ. Cardiovasc. Imaging 2016, 9, e005066. [Google Scholar] [CrossRef] [Green Version]
- Castaño, A.; Narotsky, D.L.; Hamid, N.; Khalique, O.K.; Morgenstern, R.; DeLuca, A.; Rubin, J.; Chiuzan, C.; Nazif, T.; Vahl, T.; et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur. Heart J. 2017, 38, 2879–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ternacle, J.; Krapf, L.; Mohty, D.; Magne, J.; Nguyen, A.; Galat, A.; Gallet, R.; Teiger, E.; Côté, N.; Clavel, M.A.; et al. Aortic Stenosis and Cardiac Amyloidosis: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 2638–2651. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, N.; Rella, V.; Fumagalli, C.; Salerno, S.; Castelletti, S.; Dagradi, F.; Torchio, M.; Marceca, A.; Meda, M.; Gasparini, M.; et al. Prevalence of cardiac amyloidosis among adult patients referred to tertiary centres with an initial diagnosis of hypertrophic cardiomyopathy. Int. J. Cardiol. 2020, 300, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Saelices, L.; Johnson, L.M.; Liang, W.Y.; Sawaya, M.R.; Cascio, D.; Ruchala, P.; Whitelegge, J.; Jiang, L.; Riek, R.; Eisenberg, D.S. Uncovering the Mechanism of Aggregation of Human Transthyretin. J. Biol. Chem. 2015, 290, 28932–28943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulawa, C.E.; Connelly, S.; Devit, M.; Wang, L.; Weigel, C.; Fleming, J.A.; Packman, J.; Powers, E.T.; Wiseman, R.L.; Foss, T.R.; et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. USA 2012, 109, 9629–9634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Hu, C.; Dong, Y.; Lin, M.S.; Liu, J.; Jiang, X.; Ge, Y.; Guo, Y. The impact of V30A mutation on transthyretin protein structural stability and cytotoxicity against neuroblastoma cells. Arch. Biochem. Biophys. 2013, 535, 120–127. [Google Scholar] [CrossRef]
- Wieczorek, E.; Ożyhar, A. Transthyretin: From Structural Stability to Osteoarticular and Cardiovascular Diseases. Cells 2021, 10, 1768. [Google Scholar] [CrossRef]
- Macedo, A.V.S.; Schwartzmann, P.V.; de Gusmão, B.M.; Melo, M.D.T.; Coelho-Filho, O.R. Advances in the Treatment of Cardiac Amyloidosis. Curr. Treat. Options Oncol. 2020, 21, 36. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, S.; Imai, E.; Horio, M.; Yasuda, Y.; Tomita, K.; Nitta, K.; Yamagata, K.; Tomino, Y.; Yokoyama, H.; Hishida, A. Revised equations for estimated GFR from serum creatinine in Japan. Am. J. Kidney Dis. 2009, 53, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2015, 28, 1–39.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustavsson, A.; Engström, U.; Westermark, P. Mechanisms of transthyretin amyloidogenesis. Antigenic mapping of transthyretin purified from plasma and amyloid fibrils and within in situ tissue localizations. Am. J. Pathol. 1994, 144, 1301–1311. [Google Scholar]
- Hoshii, Y.; Setoguchi, M.; Iwata, T.; Ueda, J.; Cui, D.; Kawano, H.; Gondo, T.; Takahashi, M.; Ishihara, T. Useful polyclonal antibodies against synthetic peptides corresponding to immunoglobulin light chain constant region for immunohistochemical detection of immunoglobulin light chain amyloidosis. Pathol. Int. 2001, 51, 264–270. [Google Scholar] [CrossRef]
- Cueto-Garcia, L.; Tajik, A.J.; Kyle, R.A.; Edwards, W.D.; Greipp, P.R.; Callahan, J.A.; Shub, C.; Seward, J.B. Serial echocardiographic observations in patients with primary systemic amyloidosis: An introduction to the concept of early (asymptomatic) amyloid infiltration of the heart. Mayo Clin. Proc. 1984, 59, 589–597. [Google Scholar] [CrossRef]
- Lai, H.J.; Huang, K.C.; Liang, Y.C.; Chien, K.L.; Lee, M.J.; Hsieh, S.T.; Chao, C.C.; Yang, C.C. Cardiac manifestations and prognostic implications of hereditary transthyretin amyloidosis associated with transthyretin Ala97Ser. J. Med. Assoc. 2020, 119, 693–700. [Google Scholar] [CrossRef]
- Castiglione, V.; Franzini, M.; Aimo, A.; Carecci, A.; Lombardi, C.M.; Passino, C.; Rapezzi, C.; Emdin, M.; Vergaro, G. Use of biomarkers to diagnose and manage cardiac amyloidosis. Eur. J. Heart Fail. 2021, 23, 217–230. [Google Scholar] [CrossRef]
- Pucci, A.; Aimo, A.; Musetti, V.; Barison, A.; Vergaro, G.; Genovesi, D.; Giorgetti, A.; Masotti, S.; Arzilli, C.; Prontera, C.; et al. Amyloid Deposits and Fibrosis on Left Ventricular Endomyocardial Biopsy Correlate With Extracellular Volume in Cardiac Amyloidosis. J. Am. Heart Assoc. 2021, 10, e020358. [Google Scholar] [CrossRef]
- Misumi, Y.; Ando, Y.; Gonçalves, N.P.; Saraiva, M.J. Fibroblasts endocytose and degrade transthyretin aggregates in transthyretin-related amyloidosis. Lab. Invest. 2013, 93, 911–920. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Yang, Z.G.; Wen, L.Y.; Liu, X.; Xu, H.Y.; Zhang, Q.; Guo, Y.K. Regional myocardial microvascular dysfunction in cardiac amyloid light-chain amyloidosis: Assessment with 3T cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2016, 18, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, M.M.; Saraiva, M.J. Neurodegeneration in familial amyloid polyneuropathy: From pathology to molecular signaling. Prog. Neurobiol. 2003, 71, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Sartiani, L.; Bucciantini, M.; Spinelli, V.; Leri, M.; Natalello, A.; Nosi, D.; Maria Doglia, S.; Relini, A.; Penco, A.; Giorgetti, S.; et al. Biochemical and Electrophysiological Modification of Amyloid Transthyretin on Cardiomyocytes. Biophys. J. 2016, 111, 2024–2038. [Google Scholar] [CrossRef] [Green Version]
- Sapp, V.; Jain, M.; Liao, R. Viewing Extrinsic Proteotoxic Stress Through the Lens of Amyloid Cardiomyopathy. Physiology 2016, 31, 294–299. [Google Scholar] [CrossRef]
- Griffin, J.M.; Rosenblum, H.; Maurer, M.S. Pathophysiology and Therapeutic Approaches to Cardiac Amyloidosis. Circ. Res. 2021, 128, 1554–1575. [Google Scholar] [CrossRef] [PubMed]
- Martins, D.; Moreira, J.; Gonçalves, N.P.; Saraiva, M.J. MMP-14 overexpression correlates with the neurodegenerative process in familial amyloidotic polyneuropathy. Dis. Model Mech. 2017, 10, 1253–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, P.F.; Cerca, F.; Santos, S.D.; Saraiva, M.J. Endoplasmic reticulum stress associated with extracellular aggregates. J. Biol. Chem. 2006, 281, 21998–22003. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, E.P.; Guimaraes-Costa, A.B.; Bandeira-Melo, C.; Chimelli, L.; Waddington-Cruz, M.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. Inflammatory profiling of patients with familial amyloid polyneuropathy. BMC Neurol. 2019, 19, 146–152. [Google Scholar] [CrossRef]
- Fong, V.H.; Vieira, A. Cytotoxic Effects of Transthyretin Aggregates in an Epidermoid Cell Line. Pathobiology 2017, 84, 218–222. [Google Scholar] [CrossRef]
- Manral, P.; Reixach, N. Amyloidogenic and non-amyloidogenic transthyretin variants interact differently with human cardiomyocytes: Insights into early events of non-fibrillar tissue damage. Biosci. Rep. 2015, 35, e00172. [Google Scholar] [CrossRef]
- Aimo, A.; Castiglione, V.; Borrelli, C.; Saccaro, L.F.; Franzini, M.; Masi, S.; Emdin, M.; Giannoni, A. Oxidative stress and inflammation in the evolution of heart failure: From pathophysiology to therapeutic strategies. Eur. J. Prev. Cardiol. 2020, 27, 494–510. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R.; Marbán, E. Molecular and cellular mechanisms of myocardial stunning. Physiol. Rev. 1999, 79, 609–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolli, R. Mechanism of myocardial “stunning”. Circulation 1990, 82, 723–738. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.; Gwathmey, J.K.; Xie, L.H. Oxidative stress-mediated effects of angiotensin II in the cardiovascular system. World J. Hypertens. 2012, 2, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Vergaro, G.; Aimo, A.; Campora, A.; Castiglione, V.; Prontera, C.; Masotti, S.; Musetti, V.; Chianca, M.; Valleggi, A.; Spini, V.; et al. Patients with cardiac amyloidosis have a greater neurohormonal activation than those with non-amyloidotic heart failure. Amyloid 2021, 28, 252–258. [Google Scholar] [CrossRef]
- Puig-Carrion, G.D.; Reyentovich, A.; Katz, S.D. Diagnosis and treatment of heart failure in hereditary transthyretin amyloidosis. Clin. Auton. Res. 2019, 29, 45–53. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | ATTR-CM (n = 28) |
|---|---|
| Age, years | 76.2 ± 6.4 |
| Male sex, n (%) | 26 (92.9%) |
| Comorbidities | |
| Hypertension, n (%) | 16 (57.1%) |
| Diabetes Mellitus, n (%) | 10 (35.7%) |
| Oral Medications | |
| A CE-I/ARB, n (%) | 18 (64.3%) |
| β-blocker, n (%) | 13 (46.4%) |
| Loop diuretic, n (%) | 17 (60.7%) |
| Mineralocorticoid receptor antagonists, n (%) | 10 (35.7%) |
| Electrocardiography | |
| Low QRS voltage in limb leads, n (%) | 16 (57.1%) |
| Poor R-wave progression in precordial leads, n (%) | 15 (53.6%) |
| Atrial fibrillation, n (%) | 13 (46.4%) |
| Patterns of LV geometry | |
| concentric LV hypertrophy, n (%) | 27 (96.4%) |
| concentric LV remodeling, n (%) | 1 (3.6%) |
| Transthoracic echocardiography | |
| LV ejection fraction, % | 52.4 ± 10.6 |
| LV end-diastolic diameter, mm | 44.5 ± 5.6 |
| LV end-systolic diameter, mm | 34.7 ± 9.4 |
| Intraventricular septal thickness, mm | 15.6 ± 2.7 |
| Posterior wall thickness, mm | 15.1 ± 2.3 |
| Relative wall thickness | 0.69 ± 0.13 |
| LV mass index, g/m2 | 175.6 ± 45.5 |
| E/e’ (septal) ratio | 21.7 ± 7.1 |
| Laboratory parameters | |
| Hemoglobin, g/dL | 12.7 ± 2.1 |
| Uric acid, mg/dL | 6.4 ± 2.1 |
| eGFR, mL/min/1.73 m2 | 47.3 ± 15.7 |
| Sodium, mEq/L | 139.4 ± 2.3 |
| Potassium, mEq/L | 4.4 ± 0.5 |
| Brain natriuretic peptide, pg/mL | 298.1 (202.2, 533.9) |
| High-sensitivity cardiac troponin T, ng/L | 72.5 (41.5, 95.0) |
| Plasma renin activity, µg/mL/h * | 5.4 (4.1, 10.0) |
| Plasma aldosterone concentration, pg/mL † | 200.7 ± 183.6 |
| Characteristic | ATTR-CM (n = 28) |
|---|---|
| Percentage of the TTR area | 36.2 ± 16.4 (%) |
| Percentage of the non-cardiomyocyte area | 60.2 ± 20.5 (%) |
| Percentage of the cardiomyocyte area | 39.8 ± 20.5 (%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakano, T.; Onoue, K.; Terada, C.; Terasaki, S.; Ishihara, S.; Hashimoto, Y.; Nakada, Y.; Nakagawa, H.; Ueda, T.; Seno, A.; et al. Transthyretin Amyloid Cardiomyopathy: Impact of Transthyretin Amyloid Deposition in Myocardium on Cardiac Morphology and Function. J. Pers. Med. 2022, 12, 792. https://doi.org/10.3390/jpm12050792
Nakano T, Onoue K, Terada C, Terasaki S, Ishihara S, Hashimoto Y, Nakada Y, Nakagawa H, Ueda T, Seno A, et al. Transthyretin Amyloid Cardiomyopathy: Impact of Transthyretin Amyloid Deposition in Myocardium on Cardiac Morphology and Function. Journal of Personalized Medicine. 2022; 12(5):792. https://doi.org/10.3390/jpm12050792
Chicago/Turabian StyleNakano, Tomoya, Kenji Onoue, Chiyoko Terada, Satoshi Terasaki, Satomi Ishihara, Yukihiro Hashimoto, Yasuki Nakada, Hitoshi Nakagawa, Tomoya Ueda, Ayako Seno, and et al. 2022. "Transthyretin Amyloid Cardiomyopathy: Impact of Transthyretin Amyloid Deposition in Myocardium on Cardiac Morphology and Function" Journal of Personalized Medicine 12, no. 5: 792. https://doi.org/10.3390/jpm12050792
APA StyleNakano, T., Onoue, K., Terada, C., Terasaki, S., Ishihara, S., Hashimoto, Y., Nakada, Y., Nakagawa, H., Ueda, T., Seno, A., Nishida, T., Watanabe, M., Hoshii, Y., Hatakeyama, K., Sakaguchi, Y., Ohbayashi, C., & Saito, Y. (2022). Transthyretin Amyloid Cardiomyopathy: Impact of Transthyretin Amyloid Deposition in Myocardium on Cardiac Morphology and Function. Journal of Personalized Medicine, 12(5), 792. https://doi.org/10.3390/jpm12050792