
MDMF: Predicting miRNA–Disease Association Based on Matrix Factorization with Disease Similarity Constraint


Abstract
:1. Introduction
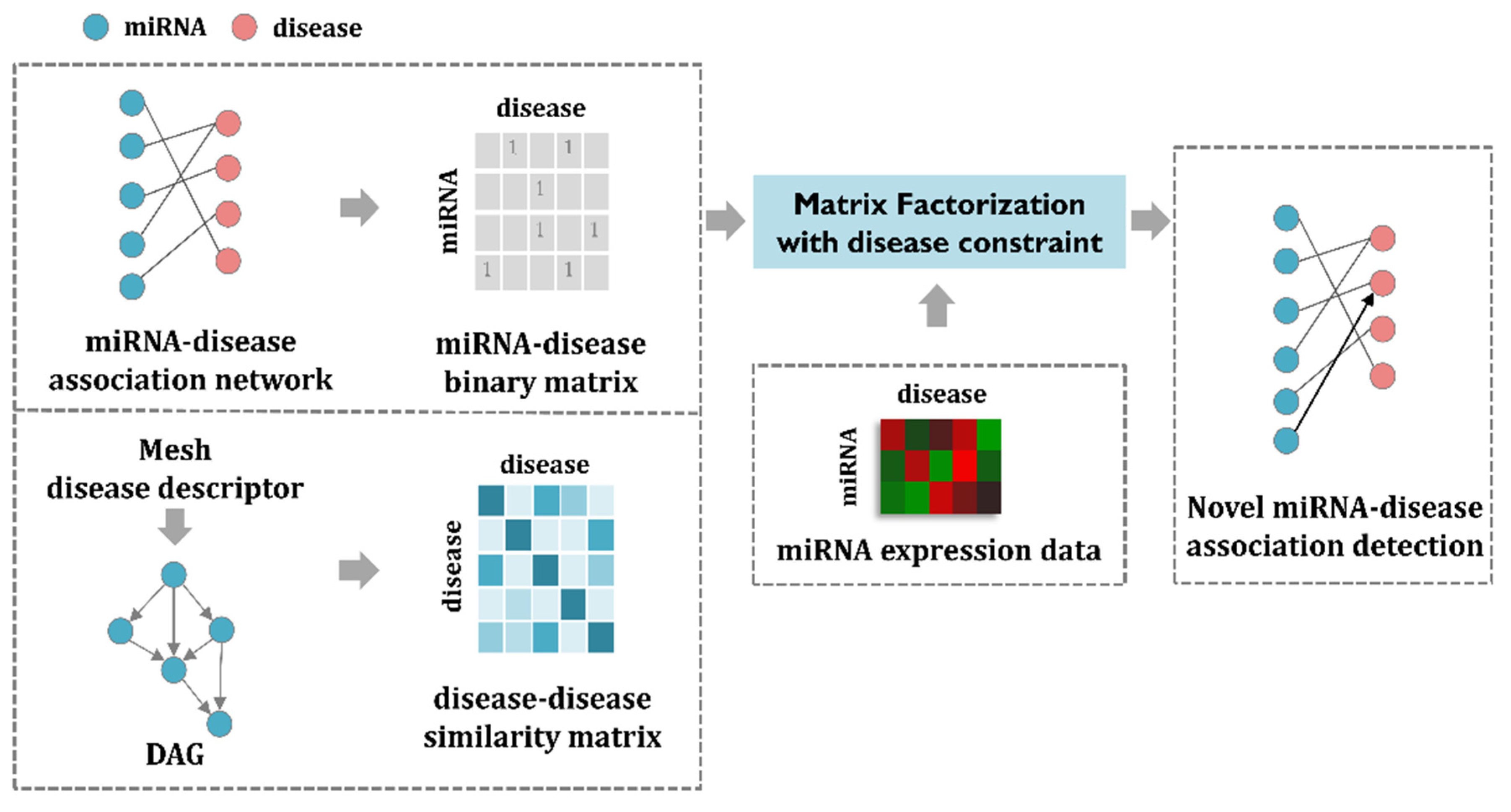
2. Materials and Methods
2.1. Human miRNA–Disease Association Data
2.2. miRNA Expression Data
2.3. Disease Semantic Similarity
2.4. Gaussian Interaction Profile Kernel Disease Similarity
2.5. Integrated Disease Similarity
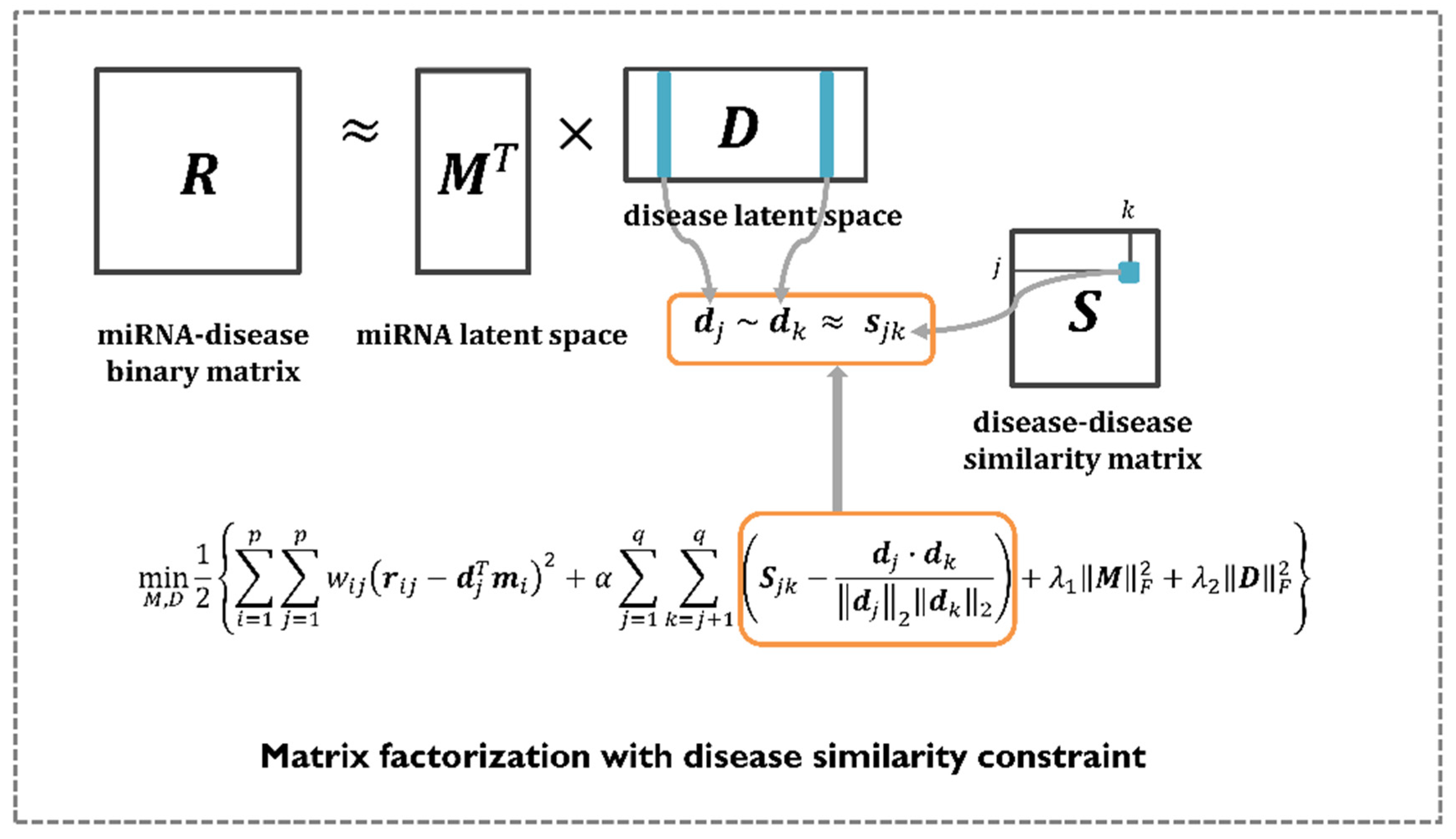
2.6. EMDMF
3. Results
3.1. Evaluation Metric
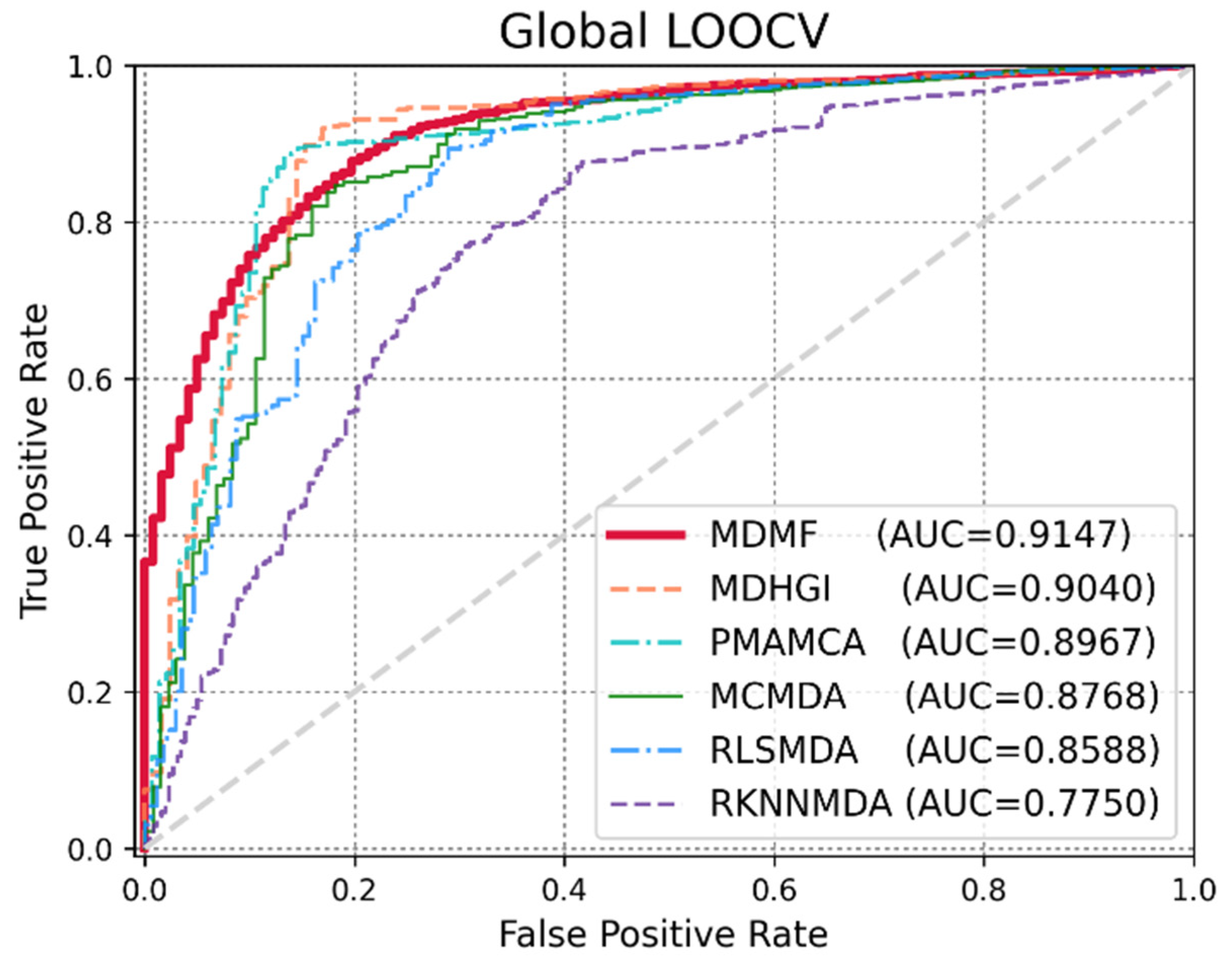
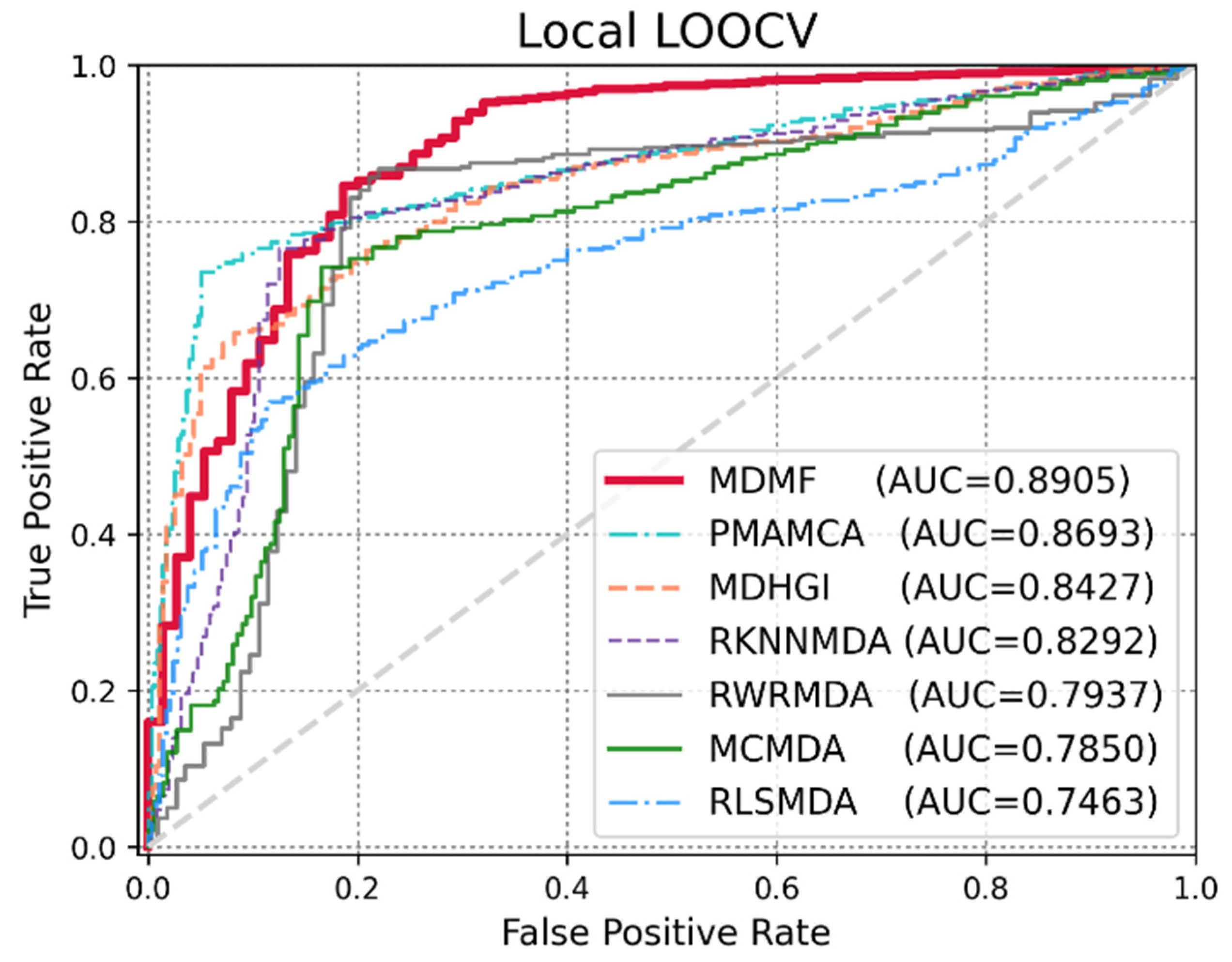
3.2. Performance Comparison with Previous Methods
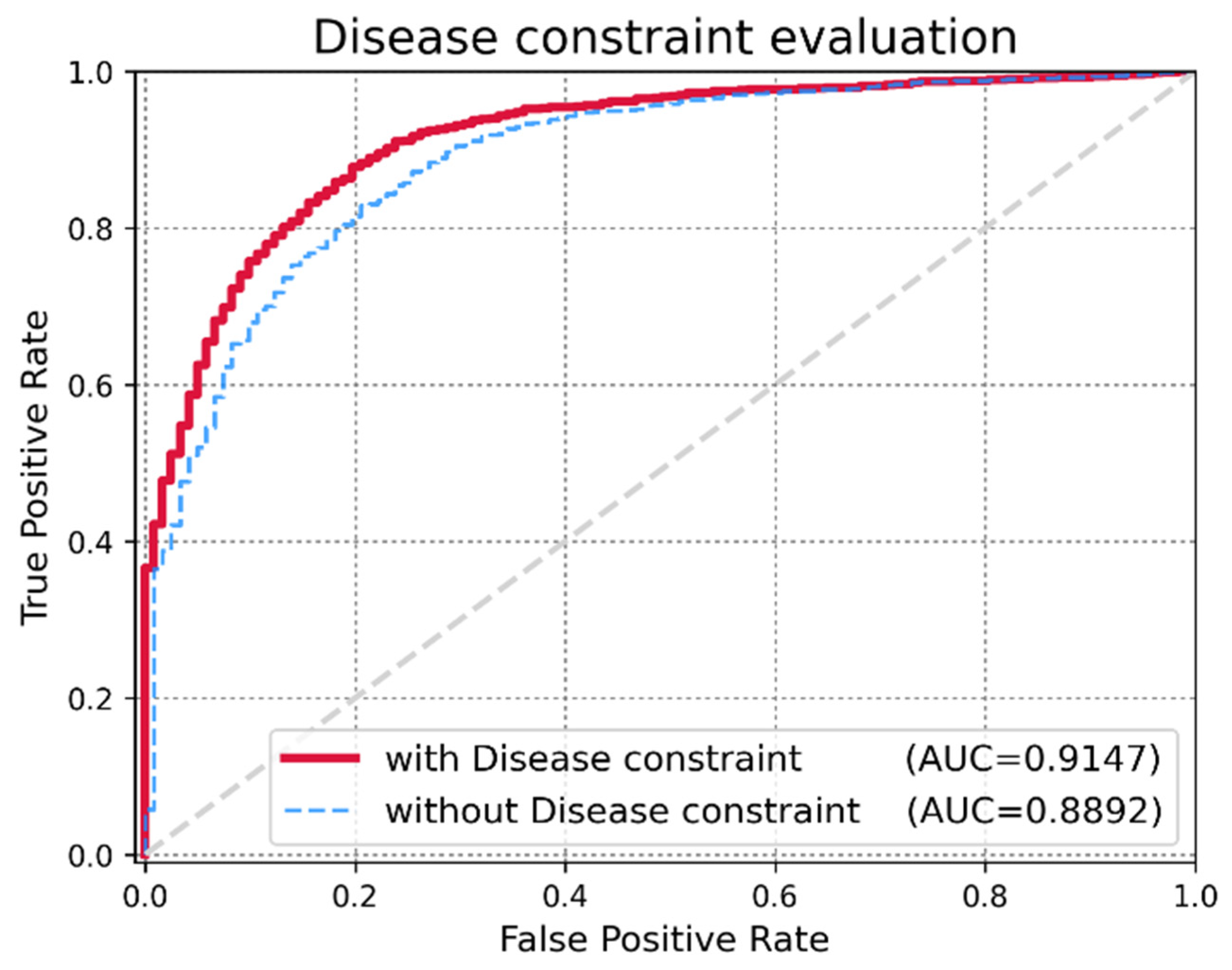
3.3. Effect of Disease Similarity Constraint
3.4. Case Studies (Breast Cancer and Lung Cancer)
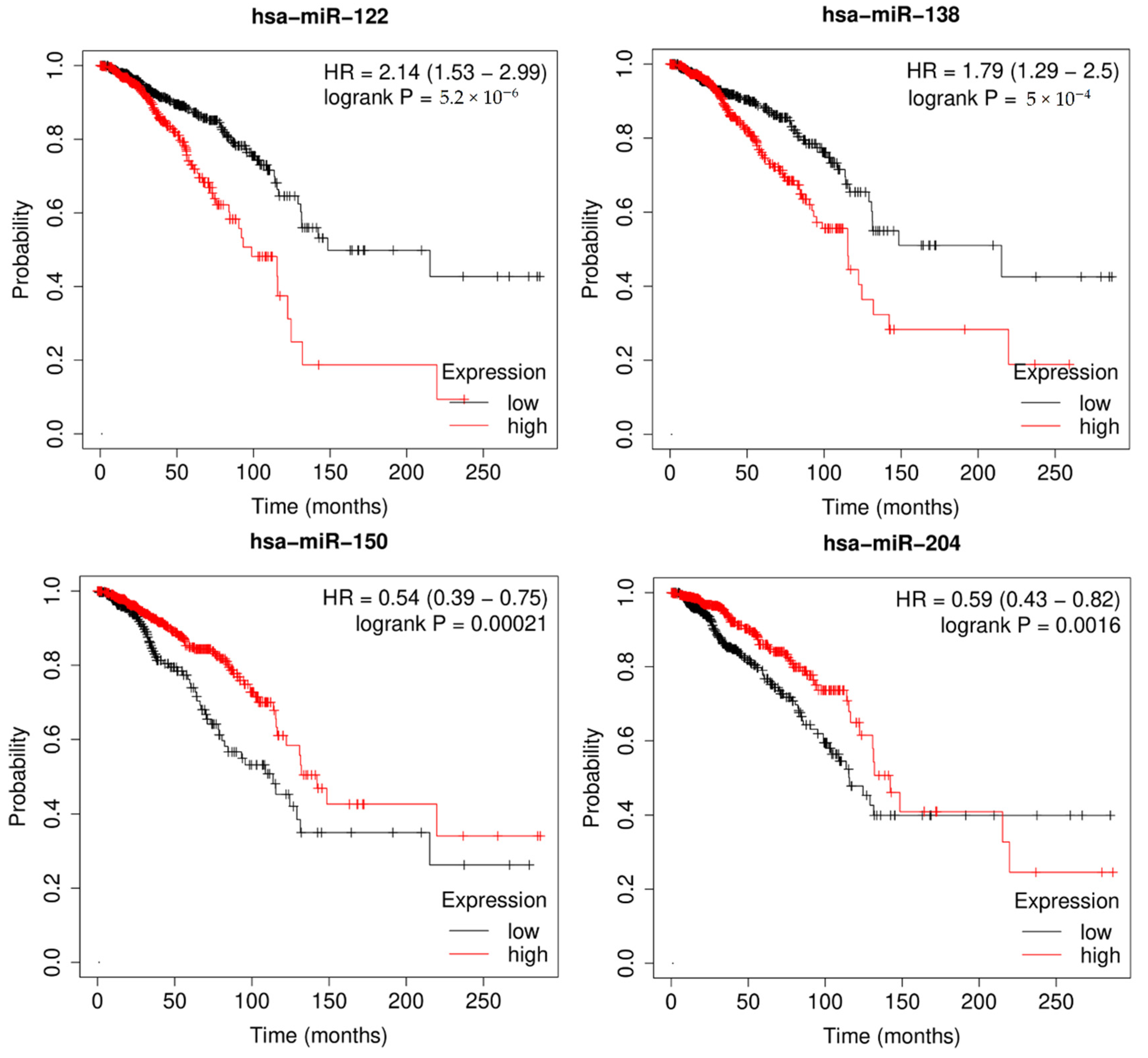
3.5. Survival Analysis
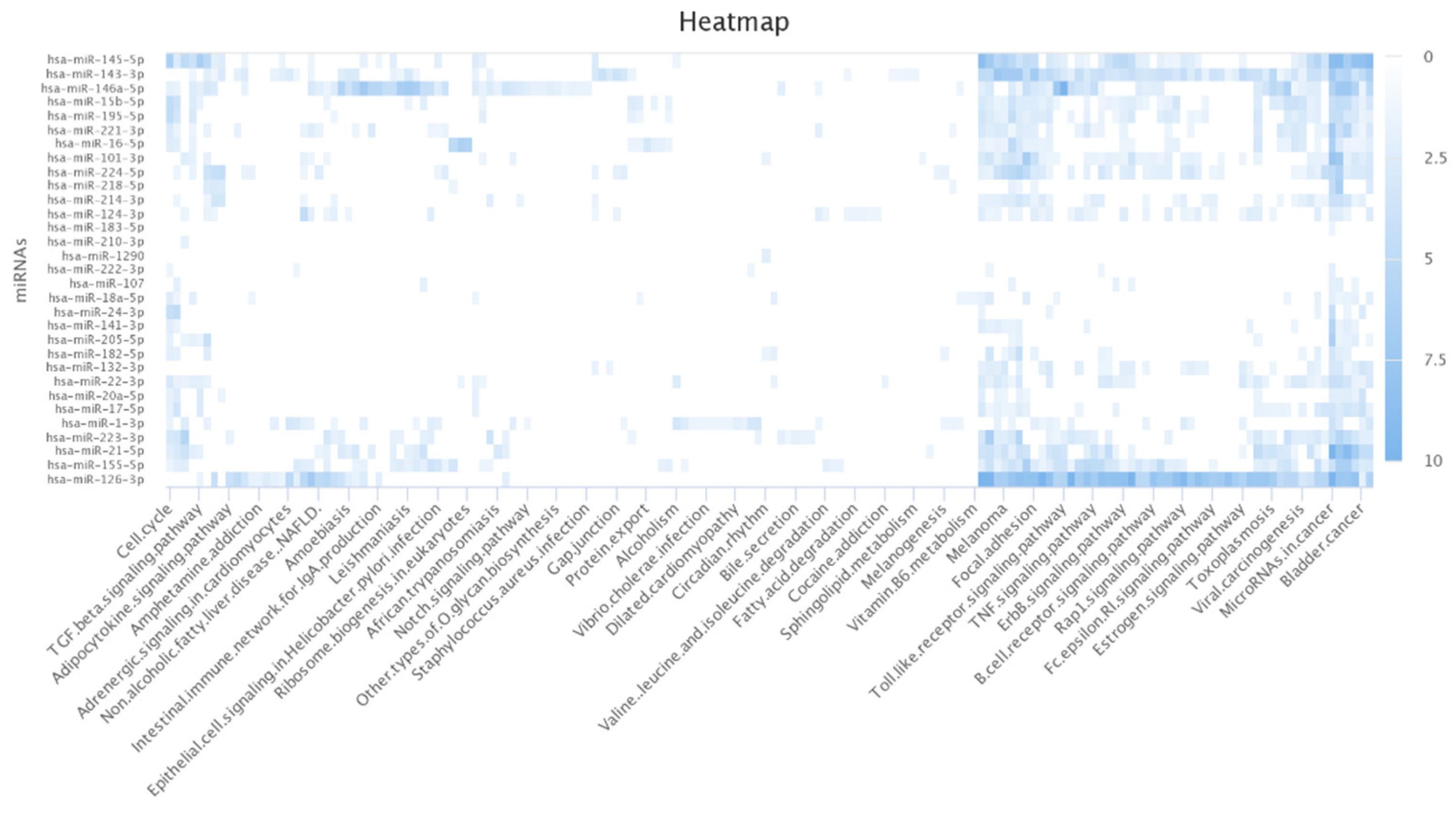
3.6. Pathway Analysis
4. Conclusions and Future Perspective
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: microRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Guo, M.; Hay, B.A. MicroRNAs and the regulation of cell death. Trends Genet. 2004, 20, 617–624. [Google Scholar] [CrossRef]
- Karp, X.; Ambros, V. Developmental biology. Encountering microRNAs in cell fate signaling. Science 2005, 310, 1288–1289. [Google Scholar] [CrossRef] [Green Version]
- Miska, E.A. How microRNAs control cell division, differentiation and death. Curr. Opin. Genet. Dev. 2005, 15, 563–568. [Google Scholar] [CrossRef]
- Cheng, A.M.; Byrom, M.W.; Shelton, J.; Ford, L.P. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005, 33, 1290–1297. [Google Scholar] [CrossRef] [Green Version]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Garcia, I.; Miska, E.A. MicroRNA functions in animal development and human disease. Development 2005, 132, 4653–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Png, K.J.; Yoshida, M.; Zhang, X.H.-F.; Shu, W.; Lee, H.; Rimner, A.; Chan, T.A.; Comen, E.; Andrade, V.P.; Kim, S.W.; et al. MicroRNA-335 inhibits tumor reinitiation and is silenced through genetic and epigenetic mechanisms in human breast cancer. Genes Dev. 2011, 25, 226–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavazoie, S.F.; Alarcón, C.; Oskarsson, T.; Padua, D.; Wang, Q.; Bos, P.D.; Gerald, W.L.; Massagué, J. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 2008, 451, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valastyan, S.; Reinhardt, F.; Benaich, N.; Calogrias, D.; Szasz, A.M.; Wang, Z.C.; Brock, J.E.; Richardson, A.L.; Weinberg, R.A. A pleiotropically acting microRNA, miR-31, inhibits breast cancer metastasis. Cell 2009, 137, 1032–1046. [Google Scholar] [CrossRef] [Green Version]
- Xuan, P.; Han, K.; Guo, M.; Guo, Y.; Li, J.; Ding, J.; Liu, Y.; Dai, Q.; Li, J.; Teng, Z.; et al. Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. PLoS ONE 2013, 8, e70204. [Google Scholar] [CrossRef]
- Jiang, Q.; Hao, Y.; Wang, G.; Juan, L.; Zhang, T.; Teng, M.; Liu, Y.; Wang, Y. Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst. Biol. 2010, 4, S2. [Google Scholar] [CrossRef] [Green Version]
- Mørk, S.; Pletscher-Frankild, S.; Palleja Caro, A.; Gorodkin, J.; Jensen, L.J. Protein-driven inference of miRNA-disease associations. Bioinformatics 2014, 30, 392–397. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, M.X.; Yan, G.Y. RWRMDA: Predicting novel human microRNA-disease associations. Mol. Biosyst. 2012, 8, 2792–2798. [Google Scholar] [CrossRef]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.; Deng, L.; Liu, Y.; Zhang, Y.; Dai, Q. WBSMDA: Within and Between Score for MiRNA-Disease Association prediction. Sci. Rep. 2016, 6, 21106. [Google Scholar] [CrossRef]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.; Huang, Y.-A.; Yan, G.-Y. HGIMDA: Heterogeneous graph inference for miRNA-disease association prediction. Oncotarget 2016, 7, 65257–65269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Xu, J.; Zhang, G.; Xu, L.; Li, C.; Wang, L.; Zhao, Z.; Jiang, W.; Guo, Z.; Li, X. Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. BMC Syst. Biol. 2013, 7, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, J.; Kim, H.; Yoon, Y.; Park, S. A method of extracting disease-related microRNAs through the propagation algorithm using the environmental factor based global miRNA network. Biomed. Mater. Eng. 2015, 26 (Suppl. S1), S1763–S1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elfaki, I.; Mir, R.; Mir, M.M.; AbuDuhier, F.M.; Babakr, A.T.; Barnawi, J. Potential impact of microRNA gene polymorphisms in the pathogenesis of diabetes and atherosclerotic cardiovascular disease. J. Pers. Med. 2019, 9, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinina, T.; Kononchuk, V.; Alekseenok, E.; Abdullin, G.; Sidorov, S.; Ovchinnikov, V.; Gulyaeva, L. Associations between the Levels of Estradiol-, Progesterone-, and Testosterone-Sensitive MiRNAs and Main Clinicopathologic Features of Breast Cancer. J. Pers. Med. 2021, 12, 4. [Google Scholar] [CrossRef]
- Izzotti, A.; Vargas, G.C.; Pulliero, A.; Coco, S.; Vanni, I.; Colarossi, C.; Blanco, G.; Agodi, A.; Barchitta, M.; Maugeri, A.; et al. Relationship between the miRNA profiles and oncogene mutations in non-smoker lung cancer. Relevance for lung cancer personalized screenings and treatments. J. Pers. Med. 2021, 11, 182. [Google Scholar] [CrossRef]
- Ha, J.; Park, C. MLMD: Metric learning for predicting MiRNA-disease associations. IEEE Access 2021, 9, 78847–78858. [Google Scholar] [CrossRef]
- Ha, J.; Park, C.; Park, S. PMAMCA: Prediction of microRNA-disease association utilizing a matrix completion approach. BMC Syst. Biol. 2019, 13, 33. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Yan, G.Y. Semi-supervised learning for potential human microRNA-disease associations inference. Sci. Rep. 2014, 4, 5501. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Yan, C.C.; Zhang, X.; Li, Z.; Deng, L.; Zhang, Y.; Dai, Q. RBMMMDA: Predicting multiple typesof disease-microRNA associations. Sci. Rep. 2015, 5, 13877. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-Q.; Rong, Z.-H.; Chen, X.; Yan, G.-Y.; You, Z. MCMDA: Matrix completion for MiRNA-disease association prediction. Oncotarget 2017, 8, 21187–21199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Luo, J.; Liang, C.; Cai, J.; Ding, P. A graph regularized non-negative matrix factorization method for identifying microRNA-disease associations. Bioinformatics 2018, 34, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, L.; Qu, J.; Guan, N.; Li, J.-Q. Predicting miRNA–disease association based on inductive matrix completion. Bioinformatics 2018, 34, 4256–4265. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yin, J.; Qu, J.; Huang, L. MDHGI: Matrix Decomposition and Heterogeneous Graph Inference for miRNA-disease association prediction. PLoS Comput. Biol. 2018, 14, e1006418. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, Q.-F.; Yan, G.-Y. RKNNMDA: Ranking-based KNN for MiRNA-Disease Association prediction. RNA Biol. 2017, 14, 952–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, J.; Park, C.; Park, C.; Park, S. IMIPMF: Inferring miRNA-disease interactions using probabilistic matrix factorization. J. Biomed. Inform. 2020, 102, 103358. [Google Scholar] [CrossRef]
- Ha, J.; Park, C.; Park, C.; Park, S. Improved Prediction of miRNA-Disease Associations Based on Matrix Completion with Network Regularization. Cells 2020, 9, 881. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Qiu, C.; Tu, J.; Geng, B.; Yang, J.; Jiang, T.; Cui, Q. HMDD v2.0: A database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014, 42, 1070–1074. [Google Scholar] [CrossRef]
- Yang, Z.; Ren, F.; Liu, C.; He, S.; Sun, G.; Gao, Q.; Yao, L.; Zhang, Y.; Miao, R.; Cao, Y.; et al. dbDEMC: A database of differentially expressed miRNAs in human cancers. BMC Genom. 2010, 11 (Suppl. S4), S5. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. miR2Disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009, 37, D98–D104. [Google Scholar] [CrossRef] [Green Version]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Współczesna Onkol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipscomb, C.E. Medical subject headings (MeSH). Bull. Med. Libr. Assoc. 2000, 88, 265–266. [Google Scholar] [PubMed]
- Wong, T.T. Performance evaluation of classification algorithms by k-fold and leave-one-out cross validation. Pattern Recognit. 2015, 48, 2839–2846. [Google Scholar] [CrossRef]
- Huang, J.; Ling, C.X. Using AUC and accuracy in evaluating learning algorithms. IEEE Trans. Knowl. Data Eng. 2005, 17, 299–310. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Sauer, A.G.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [Green Version]
- Lánczky, A.; Nagy, Á.; Bottai, G.; Munkácsy, G.; Szabó, A.; Santarpia, L.; Győrffy, B. miRpower: A web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res. Treat. 2016, 160, 439–446. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef]
- Van Meerbeeck, J.P.; Fennell, D.A.; De Ruysscher, D.K. Small-cell lung cancer. Lancet 2011, 378, 1741–1755. [Google Scholar] [CrossRef]
- Volm, M.; Koomägi, R. Hypoxia-inducible factor (HIF-1) and its relationship to apoptosis and proliferation in lung cancer. Anticancer. Res. 2000, 20, 1527–1533. [Google Scholar]
- Kehl, T.; Kern, F.; Backes, C.; Fehlmann, T.; Stöckel, D.; Meese, E.; Lenhof, H.-P.; Keller, A. miRPathDB 2.0: A novel release of the miRNA Pathway Dictionary Database. Nucleic Acids Res. 2020, 48, D142–D147. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Description |
|---|---|
| p, q, k | Number of miRNAs, diseases and latent dimensions |
| M, D | miRNA and disease latent matrix |
| S | Disease similarity matrix |
| W | miRNA similarity matrix |
| L | Objective function |
| λ | Hyper parameter for regularization |
| η | Learning rate |
| Methods | AUC | AUPR | F1 | ACC | MCC |
|---|---|---|---|---|---|
| MDMF | 0.9147 | 0.8408 | 0.8482 | 0.8547 | 71.54 |
| MDHGI | 0.9040 | 0.8404 | 0.8745 | 0.8391 | 64.49 |
| PMAMCA | 0.8967 | 0.8501 | 0.8802 | 0.8446 | 68.71 |
| MCMDA | 0.8768 | 0.8043 | 0.8704 | 0.8342 | 63.48 |
| RLSMDA | 0.8588 | 0.7647 | 0.7342 | 0.8164 | 65.42 |
| RKNNMDA | 0.7750 | 0.8482 | 0.8703 | 0.8128 | 64.81 |
| Methods | AUC | AUPR | F1 | ACC | MCC |
|---|---|---|---|---|---|
| MDMF | 0.8905 | 0.8129 | 0.8347 | 0.8538 | 69.84 |
| PMAMCA | 0.8693 | 0.8846 | 0.8284 | 0.8404 | 62.49 |
| MDHGI | 0.8427 | 0.8104 | 0.8591 | 0.8349 | 66.91 |
| RKNNMDA | 0.8292 | 0.8864 | 0.8028 | 0.8116 | 64.82 |
| RWRMDA | 0.7937 | 0.7372 | 0.7729 | 0.7527 | 59.42 |
| MCMDA | 0.7850 | 0.8764 | 0.8418 | 0.8268 | 68.16 |
| RLSMDA | 0.7463 | 0.8648 | 0.8045 | 0.8143 | 62.72 |
| α | AUC (Global LOOCV) | AUC (Local LOOCV) |
|---|---|---|
| 0.1 | 0.8874 | 0.8711 |
| 0.2 | 0.8954 | 0.8724 |
| 0.3 | 0.9018 | 0.8769 |
| 0.4 | 0.9082 | 0.8784 |
| 0.5 | 0.9127 | 0.8891 |
| 0.6 | 0.9116 | 0.8842 |
| 0.7 | 0.9147 | 0.8905 |
| 0.8 | 0.9128 | 0.8874 |
| 0.9 | 0.9042 | 0.8842 |
| Rank | Name | Evidence | Rank | Name | Evidence |
|---|---|---|---|---|---|
| 1 | hsa-miR-214 | hmdd, dbDEMC | 26 | hsa-miR-1237 | dbDEMC |
| 2 | hsa-miR-937-3p | dbDEMC | 27 | hsa-miR-129 | hmdd, dbDEMC |
| 3 | hsa-miR-1248 | hmdd, dbDEMC | 28 | hsa-miR-340* | dbDEMC |
| 4 | hsa-miR-920 | dbDEMC | 29 | hsa-miR-16-1-3p | dbDEMC |
| 5 | hsa-miR-520e | dbDEMC | 30 | hsa-miR-302b* | dbDEMC |
| 6 | hsa-miR-593 | dbDEMC | 31 | hsa-miR-1266 | hmdd, dbDEMC |
| 7 | hsa-miR-381 | hmdd, dbDEMC | 32 | hsa-miR-1249-3p | dbDEMC |
| 8 | hsa-miR-16 | hmdd, dbDEMC | 33 | hsa-miR-1262 | dbDEMC |
| 9 | hsa-miR-502 | hmdd | 34 | hsa-miR-494-3p | dbDEMC |
| 10 | hsa-let-7g* | dbDEMC | 35 | hsa-miR-1911* | dbDEMC |
| 11 | hsa-miR-370 | hmdd, dbDEMC | 36 | hsa-miR-376b-3p | dbDEMC |
| 12 | hsa-miR-330 | dbDEMC | 37 | hsa-miR-1276 | dbDEMC |
| 13 | hsa-miR-452 | hmdd, dbDEMC | 38 | hsa-miR-331-5p | dbDEMC |
| 14 | hsa-miR-124a-3 | hmdd, miR2disease | 39 | hsa-miR-302e | dbDEMC |
| 15 | hsa-miR-410-3p | dbDEMC | 40 | hsa-miR-361-5p | dbDEMC |
| 16 | hsa-miR-500a | dbDEMC | 41 | hsa-miR-205 | hmdd, miR2disease, dbDEMC |
| 17 | hsa-miR-766-3p | dbDEMC | 42 | hsa-miR-215-5p | dbDEMC |
| 18 | hsa-miR-29a-3p | dbDEMC | 43 | hsa-miR-30b-3p | dbDEMC |
| 19 | hsa-miR-23a | hmdd, dbDEMC | 44 | hsa-miR-760 | hmdd, dbDEMC |
| 20 | hsa-miR-3653-3p | dbDEMC | 45 | hsa-miR-4458 | dbDEMC |
| 21 | hsa-miR-513b | dbDEMC | 46 | hsa-miR-30c | hmdd, dbDEMC |
| 22 | hsa-miR-125a-3p | dbDEMC | 47 | hsa-miR-3121-5p | dbDEMC |
| 23 | hsa-let-7a-2-3p | dbDEMC | 48 | hsa-miR-609 | dbDEMC |
| 24 | hsa-miR-3130-2 | hmdd | 49 | hsa-miR-21* | dbDEMC |
| 25 | hsa-miR-1272 | dbDEMC | 50 | hsa-miR-7705 | dbDEMC |
| Rank | Name | Evidence | Rank | Name | Evidence |
|---|---|---|---|---|---|
| 1 | hsa-miR-214 | hmdd, dbDEMC | 26 | hsa-miR-1237 | dbDEMC |
| 2 | hsa-miR-937-3p | dbDEMC | 27 | hsa-miR-129 | hmdd, dbDEMC |
| 3 | hsa-miR-1248 | hmdd, dbDEMC | 28 | hsa-miR-340* | dbDEMC |
| 4 | hsa-miR-920 | dbDEMC | 29 | hsa-miR-16-1-3p | dbDEMC |
| 5 | hsa-miR-520e | dbDEMC | 30 | hsa-miR-302b* | dbDEMC |
| 6 | hsa-miR-593 | dbDEMC | 31 | hsa-miR-1266 | hmdd, dbDEMC |
| 7 | hsa-miR-381 | hmdd, dbDEMC | 32 | hsa-miR-1249-3p | dbDEMC |
| 8 | hsa-miR-16 | hmdd, dbDEMC | 33 | hsa-miR-1262 | dbDEMC |
| 9 | hsa-miR-502 | hmdd | 34 | hsa-miR-494-3p | dbDEMC |
| 10 | hsa-let-7g* | dbDEMC | 35 | hsa-miR-1911* | dbDEMC |
| 11 | hsa-miR-370 | hmdd, dbDEMC | 36 | hsa-miR-376b-3p | dbDEMC |
| 12 | hsa-miR-330 | dbDEMC | 37 | hsa-miR-1276 | dbDEMC |
| 13 | hsa-miR-452 | hmdd, dbDEMC | 38 | hsa-miR-331-5p | dbDEMC |
| 14 | hsa-miR-124a-3 | hmdd, miR2disease | 39 | hsa-miR-302e | dbDEMC |
| 15 | hsa-miR-410-3p | dbDEMC | 40 | hsa-miR-361-5p | dbDEMC |
| 16 | hsa-miR-500a | dbDEMC | 41 | hsa-miR-205 | hmdd, miR2disease, dbDEMC |
| 17 | hsa-miR-766-3p | dbDEMC | 42 | hsa-miR-215-5p | dbDEMC |
| 18 | hsa-miR-29a-3p | dbDEMC | 43 | hsa-miR-30b-3p | dbDEMC |
| 19 | hsa-miR-23a | hmdd, dbDEMC | 44 | hsa-miR-760 | hmdd, dbDEMC |
| 20 | hsa-miR-3653-3p | dbDEMC | 45 | hsa-miR-4458 | dbDEMC |
| 21 | hsa-miR-513b | dbDEMC | 46 | hsa-miR-30c | hmdd, dbDEMC |
| 22 | hsa-miR-125a-3p | dbDEMC | 47 | hsa-miR-3121-5p | dbDEMC |
| 23 | hsa-let-7a-2-3p | dbDEMC | 48 | hsa-miR-609 | dbDEMC |
| 24 | hsa-miR-3130-2 | hmdd | 49 | hsa-miR-21* | dbDEMC |
| 25 | hsa-miR-1272 | dbDEMC | 50 | hsa-miR-7705 | dbDEMC |
| KEGG Pathway | p-Value |
|---|---|
| Hippo signaling pathway | 1.41440646708 × 10−7 |
| Chronic myeloid leukemia | 6.87396730677 × 10−6 |
| TGF-beta signaling pathway | 7.52715819175 × 10−6 |
| ECM-receptor interaction | 1.33810742874 × 10−5 |
| FoxO signaling pathway | 7.94489535244 × 10−5 |
| Prostate cancer | 0.00245651291245 |
| Non-small cell lung cancer (NSCLC) | 0.00329923289869 |
| Thyroid cancer | 0.00715240823084 |
| ErbB signaling pathway | 0.00817122414933 |
| Pancreatic cancer | 0.0120595309627 |
| p53 signaling pathway | 0.022215235485 |
| HIF-1 signaling pathway | 0.0429548116057 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, J. MDMF: Predicting miRNA–Disease Association Based on Matrix Factorization with Disease Similarity Constraint. J. Pers. Med. 2022, 12, 885. https://doi.org/10.3390/jpm12060885
Ha J. MDMF: Predicting miRNA–Disease Association Based on Matrix Factorization with Disease Similarity Constraint. Journal of Personalized Medicine. 2022; 12(6):885. https://doi.org/10.3390/jpm12060885
Chicago/Turabian StyleHa, Jihwan. 2022. "MDMF: Predicting miRNA–Disease Association Based on Matrix Factorization with Disease Similarity Constraint" Journal of Personalized Medicine 12, no. 6: 885. https://doi.org/10.3390/jpm12060885
APA StyleHa, J. (2022). MDMF: Predicting miRNA–Disease Association Based on Matrix Factorization with Disease Similarity Constraint. Journal of Personalized Medicine, 12(6), 885. https://doi.org/10.3390/jpm12060885