
Combining Clinical and Genetic Data to Predict Response to Fingolimod Treatment in Relapsing Remitting Multiple Sclerosis Patients: A Precision Medicine Approach

 , ,
, , 
 and
and
Abstract
:1. Introduction
1.1. Background
1.2. Rationale
1.3. Objective
2. Materials and Methods
2.1. Genotyping and Quality Checks
2.2. Predictive Models
2.2.1. Genetic Model
2.2.2. Combined Clinical and Genetic Model
2.2.3. Predictive Performance of the Genetic Model in Patients Treated with Other Immunomodulatory Drugs
2.2.4. Classification Performance of the Combined Model
3. Results
3.1. Summary of Results
3.2. Detailed Results
3.3. Genetic Model
3.4. Combined Clinical and Genetic Model
3.5. Evaluation of the Model in Independent Cohorts of Patients Treated with First-Line Drugs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple Sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Gold, R.; Thompson, A.J.; Otero-Romero, S.; Amato, M.P.; Chandraratna, D.; Clanet, M.; Comi, G.; Derfuss, T.; Fazekas, F.; et al. ECTRIMS/EAN Guideline on the Pharmacological Treatment of People with Multiple Sclerosis. Eur. J. Neurol. 2018, 25, 215–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsopoulos, N.A.; Baranzini, S.E.; Santaniello, A.; Shoostari, P.; Cotsapas, C.; Wong, G.; Beecham, A.H.; James, T.; Replogle, J.; Vlachos, I.S.; et al. Multiple Sclerosis Genomic Map Implicates Peripheral Immune Cells & Microglia in Susceptibility. Science 2019, 365, 50. [Google Scholar] [CrossRef] [Green Version]
- Hauser, S.L.; Oksenberg, J.R.; Lincoln, R.; Garovoy, J.; Beck, R.W.; Cole, S.R.; Moke, P.S.; Kip, K.E.; Gal, R.L.; Long, D.T. Interaction between HLA-DR2 and Abnormal Brain MRI in Optic Neuritis and Early MS. Neurology 2000, 54, 1859–1861. [Google Scholar] [CrossRef]
- Barcellos, L.F.; Oksenberg, J.R.; Begovich, A.B.; Martin, E.R.; Schmidt, S.; Vittinghoff, E.; Goodin, D.S.; Pelletier, D.; Lincoln, R.R.; Bucher, P.; et al. HLA-DR2 Dose Effect on Susceptibility to Multiple Sclerosis and Influence on Disease Course. Am. J. Hum. Genet. 2003, 72, 710–716. [Google Scholar] [CrossRef] [Green Version]
- Briggs, F.B.S.; Shao, X.; Goldstein, B.A.; Oksenberg, J.R.; Barcellos, L.F.; De Jager, P.L. Genome-Wide Association Study of Severity in Multiple Sclerosis. Genes Immun. 2011, 12, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Comabella, M.; Craig, D.W.; Morcillo-Suárez, C.; Río, J.; Navarro, A.; Fernández, M.; Martin, R.; Montalban, X. Genome-Wide Scan of 500 000 Single-Nucleotide Polymorphisms Among Responders and Nonresponders to Interferon Beta Therapy in Multiple Sclerosis. Arch. Neurol. 2009, 66, 972–978. [Google Scholar] [CrossRef] [Green Version]
- Esposito, F.; Sorosina, M.; Ottoboni, L.; Lim, E.T.; Replogle, J.M.; Raj, T.; Brambilla, P.; Liberatore, G.; Guaschino, C.; Romeo, M.; et al. A Pharmacogenetic Study Implicates SLC9a9 in Multiple Sclerosis Disease Activity. Ann. Neurol. 2015, 78, 115–127. [Google Scholar] [CrossRef]
- Clarelli, F.; Liberatore, G.; Sorosina, M.; Osiceanu, A.M.; Esposito, F.; Mascia, E.; Santoro, S.; Pavan, G.; Colombo, B.; Moiola, L.; et al. Pharmacogenetic Study of Long-Term Response to Interferon-β Treatment in Multiple Sclerosis. Pharm. J. 2017, 17, 84–91. [Google Scholar] [CrossRef]
- Mahurkar, S.; Moldovan, M.; Suppiah, V.; Sorosina, M.; Clarelli, F.; Liberatore, G.; Malhotra, S.; Montalban, X.; Antigüedad, A.; Krupa, M.; et al. Response to Interferon-Beta Treatment in Multiple Sclerosis Patients: A Genome-Wide Association Study. Pharm. J. 2017, 17, 312–318. [Google Scholar] [CrossRef]
- Grossman, I.; Avidan, N.; Singer, C.; Goldstaub, D.; Hayardeny, L.; Eyal, E.; Ben-Asher, E.; Paperna, T.; Pe’er, I.; Lancet, D.; et al. Pharmacogenetics of Glatiramer Acetate Therapy for Multiple Sclerosis Reveals Drug-Response Markers. Pharmacogenet. Genom. 2007, 17, 657–666. [Google Scholar] [CrossRef]
- Kulakova, O.; Bashinskaya, V.; Kiselev, I.; Baulina, N.; Tsareva, E.; Nikolaev, R.; Kozin, M.; Shchur, S.; Favorov, A.; Boyko, A.; et al. Pharmacogenetics of Glatiramer Acetate Therapy for Multiple Sclerosis: The Impact of Genome-Wide Association Studies Identified Disease Risk Loci. Pharmacogenomics 2017, 18, 1563–1574. [Google Scholar] [CrossRef]
- Tsareva, E.Y.; Kulakova, O.G.; Boyko, A.N.; Shchur, S.G.; Lvovs, D.; Favorov, A.V.; Gusev, E.I.; Vandenbroeck, K.; Favorova, O.O. Allelic Combinations of Immune-Response Genes Associated with Glatiramer Acetate Treatment Response in Russian Multiple Sclerosis Patients. Pharmacogenomics 2012, 13, 43–53. [Google Scholar] [CrossRef]
- Dominguez-Mozo, M.I.; Perez-Perez, S.; Villar, L.M.; Oliver-Martos, B.; Villarrubia, N.; Matesanz, F.; Costa-Frossard, L.; Pinto-Medel, M.J.; García-Sánchez, M.I.; Ortega-Madueño, I.; et al. Predictive Factors and Early Biomarkers of Response in Multiple Sclerosis Patients Treated with Natalizumab. Sci. Rep. 2020, 10, 14244. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Lafuente, R.; Blanco-Kelly, F.; Garcia-Montojo, M.; Martínez, A.; De Las Heras, V.; Dominguez-Mozo, M.I.; Bartolome, M.; Garcia-Martinez, A.; De la Concha, E.G.; Urcelay, E.; et al. CD46 in a Spanish Cohort of Multiple Sclerosis Patients: Genetics, MRNA Expression and Response to Interferon-Beta Treatment. Mult. Scler. 2011, 17, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Ferrè, L.; Mogavero, A.; Clarelli, F.; Moiola, L.; Sangalli, F.; Colombo, B.; Martinelli, V.; Comi, G.; Filippi, M.; Esposito, F. Early Evidence of Disease Activity during Fingolimod Predicts Medium-Term Inefficacy in Relapsing-Remitting Multiple Sclerosis. Mult. Scler. 2021, 27, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Rajkomar, A.; Dean, J.; Kohane, I. Machine Learning in Medicine. N. Engl. J. Med. 2019, 380, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.S.W.; Schierding, W.; Wake, M.; Saffery, R.; O’Sullivan, J. Machine Learning SNP Based Prediction for Precision Medicine. Front. Genet. 2019, 10, 267. [Google Scholar] [CrossRef] [Green Version]
- Esposito, F.; Ferrè, L.; Clarelli, F.; Rocca, M.A.; Sferruzza, G.; Storelli, L.; Radaelli, M.; Sangalli, F.; Moiola, L.; Colombo, B.; et al. Effectiveness and Baseline Factors Associated to Fingolimod Response in a Real-World Study on Multiple Sclerosis Patients. J. Neurol. 2018, 265, 896–905. [Google Scholar] [CrossRef]
- Jokubaitis, V.G.; Li, V.; Kalincik, T.; Izquierdo, G.; Hodgkinson, S.; Alroughani, R.; Lechner-Scott, J.; Lugaresi, A.; Duquette, P.; Girard, M.; et al. Fingolimod after Natalizumab and the Risk of Short-Term Relapse. Neurology 2014, 82, 1204–1211. [Google Scholar] [CrossRef]
- Rotstein, D.L.; Healy, B.C.; Malik, M.T.; Chitnis, T.; Weiner, H.L. Evaluation of No Evidence of Disease Activity in a 7-Year Longitudinal Multiple Sclerosis Cohort. JAMA Neurol. 2015, 72, 152. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Ambroise, C.; McLachlan, G.J. Selection Bias in Gene Extraction on the Basis of Microarray Gene-Expression Data. Proc. Natl. Acad. Sci. USA 2002, 99, 6562–6566. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, B.A.; Polley, E.C.; Briggs, F.B.S. Random Forests for Genetic Association Studies. Stat. Appl. Genet. Mol. Biol. 2011, 10, 32. [Google Scholar] [CrossRef]
- Zhuang, X.; Ye, R.; So, M.T.; Lam, W.Y.; Karim, A.; Yu, M.; Ngo, N.D.; Cherny, S.S.; Tam, P.K.H.; Garcia-Barcelo, M.M.; et al. A Random Forest-Based Framework for Genotyping and Accuracy Assessment of Copy Number Variations. NAR Genom. Bioinforma. 2020, 2, lqaa071. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Vazquez, D.; Lopez, A.M.; Amores, J. On-Board Object Detection: Multicue, Multimodal, and Multiview Random Forest of Local Experts. IEEE Trans. Cybern. 2017, 47, 3980–3990. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene Set Analysis Toolkit with Revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [Green Version]
- Río, J.; Comabella, M.; Montalban, X. Predicting Responders to Therapies for Multiple Sclerosis. Nat. Rev. Neurol. 2009, 5, 553–560. [Google Scholar] [CrossRef]
- Danelakis, A.; Theoharis, T.; Verganelakis, D.A. Survey of Automated Multiple Sclerosis Lesion Segmentation Techniques on Magnetic Resonance Imaging. Comput. Med. Imaging Graph. 2018, 70, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Gabr, R.E.; Coronado, I.; Robinson, M.; Sujit, S.J.; Datta, S.; Sun, X.; Allen, W.J.; Lublin, F.D.; Wolinsky, J.S.; Narayana, P.A. Brain and Lesion Segmentation in Multiple Sclerosis Using Fully Convolutional Neural Networks: A Large-Scale Study. Mult. Scler. 2020, 26, 1217–1226. [Google Scholar] [CrossRef]
- Eshaghi, A.; Riyahi-Alam, S.; Saeedi, R.; Roostaei, T.; Nazeri, A.; Aghsaei, A.; Doosti, R.; Ganjgahi, H.; Bodini, B.; Shakourirad, A.; et al. Classification Algorithms with Multi-Modal Data Fusion Could Accurately Distinguish Neuromyelitis Optica from Multiple Sclerosis. NeuroImage. Clin. 2015, 7, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Eshaghi, A.; Wottschel, V.; Cortese, R.; Calabrese, M.; Sahraian, M.A.; Thompson, A.J.; Alexander, D.C.; Ciccarelli, O. Gray Matter MRI Differentiates Neuromyelitis Optica from Multiple Sclerosis Using Random Forest. Neurology 2016, 87, 2463–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Lee, Y.; Kim, Y.H.; Lim, Y.M.; Lee, J.S.; Woo, J.; Jang, S.K.; Oh, Y.J.; Kim, H.W.; Lee, E.J.; et al. Deep Learning-Based Method to Differentiate Neuromyelitis Optica Spectrum Disorder From Multiple Sclerosis. Front. Neurol. 2020, 11, 599042. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.F.; Oliveira, H.; Batista, S.; Cruz, L.; Pinto, M.; Correia, I.; Martins, P.; Teixeira, C. Prediction of Disease Progression and Outcomes in Multiple Sclerosis with Machine Learning. Sci. Rep. 2020, 10, 21038. [Google Scholar] [CrossRef]
- De Brouwer, E.; Becker, T.; Moreau, Y.; Havrdova, E.K.; Trojano, M.; Eichau, S.; Ozakbas, S.; Onofrj, M.; Grammond, P.; Kuhle, J.; et al. Longitudinal Machine Learning Modeling of MS Patient Trajectories Improves Predictions of Disability Progression. Comput. Methods Programs Biomed. 2021, 208, 106180. [Google Scholar] [CrossRef]
- Ross, C.J.; Towfic, F.; Shankar, J.; Laifenfeld, D.; Thoma, M.; Davis, M.; Weiner, B.; Kusko, R.; Zeskind, B.; Knappertz, V.; et al. A Pharmacogenetic Signature of High Response to Copaxone in Late-Phase Clinical-Trial Cohorts of Multiple Sclerosis. Genome Med. 2017, 9, 50. [Google Scholar] [CrossRef] [Green Version]
- Bin Rafiq, R.; Modave, F.; Guha, S.; Albert, M.V. Validation Methods to Promote Real-World Applicability of Machine Learning in Medicine. In Proceedings of the 2020 3rd International Conference on Digital Medicine and Image Processing, Kyoto, Japan, 6–9 November 2020; pp. 13–19. [Google Scholar] [CrossRef]
- Singh, V.; Pencina, M.; Einstein, A.J.; Liang, J.X.; Berman, D.S.; Slomka, P. Impact of Train/Test Sample Regimen on Performance Estimate Stability of Machine Learning in Cardiovascular Imaging. Sci. Rep. 2021, 11, 14490. [Google Scholar] [CrossRef]
- Berisha, V.; Krantsevich, C.; Hahn, P.R.; Hahn, S.; Dasarathy, G.; Turaga, P.; Liss, J. Digital Medicine and the Curse of Dimensionality. NPJ Digit. Med. 2021, 4, 153. [Google Scholar] [CrossRef]
- Gliozzo, J.; Mesiti, M.; Notaro, M.; Petrini, A.; Patak, A.; Puertas-Gallardo, A.; Paccanaro, A.; Valentini, G.; Casiraghi, E. Heterogeneous Data Integration Methods for Patient Similarity Networks. Brief. Bioinform. 2022, 23, bbac207. [Google Scholar] [CrossRef]


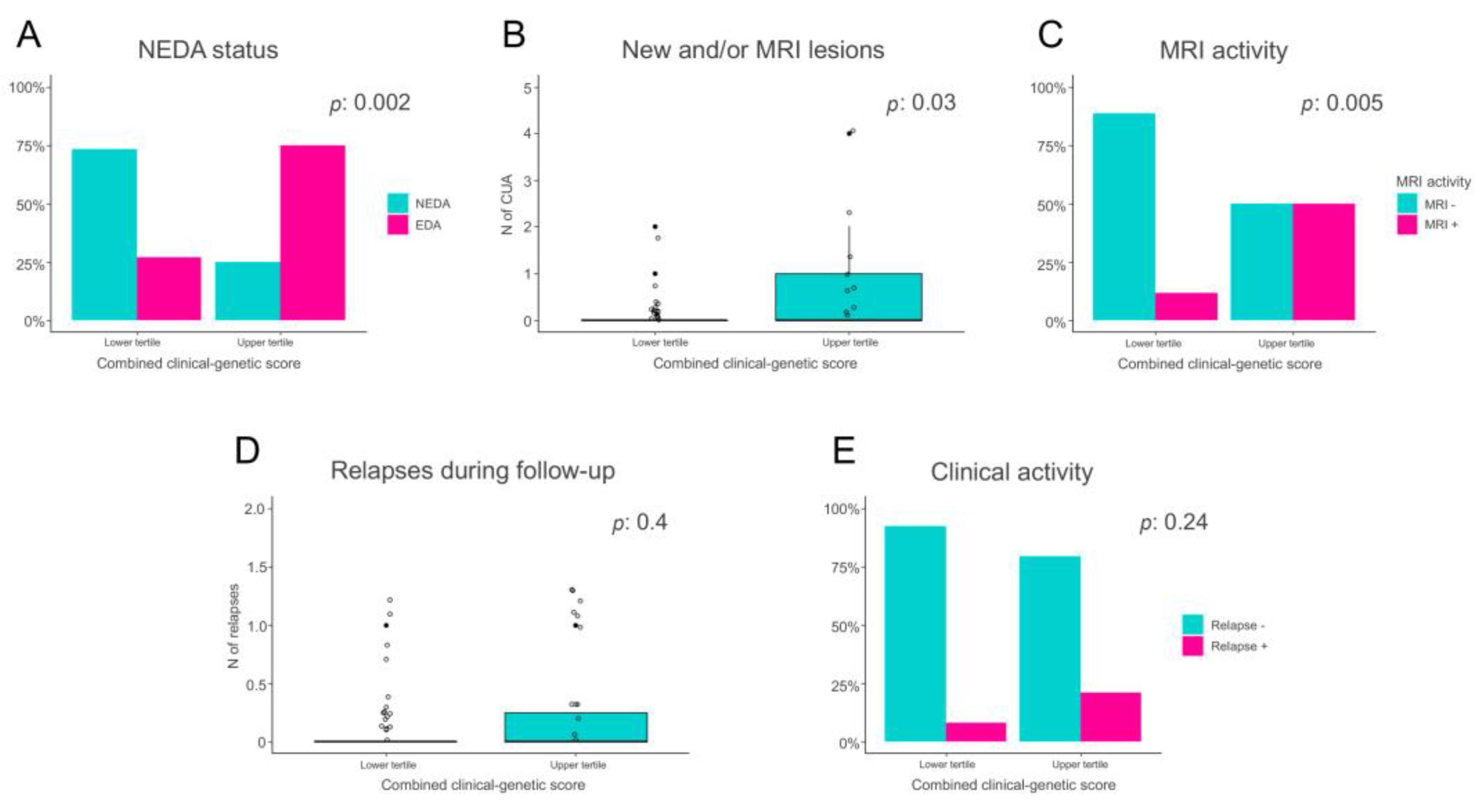{kind=link}
| Whole Cohort (n: 381) | TRset (n: 152) | Vset (n: 152) | TEset (n: 77) | p-Value | |
|---|---|---|---|---|---|
| F:M ratio | 270:111 | 101:51 | 119:33 | 50:27 | 0.03 |
| Age at disease onset, mean ± SD | 29 ± 9.5 | 28.3 ± 9.2 | 29.1 ± 9.7 | 30.2 ± 9.6 | n.s. |
| Age at FTY start, mean ± SD | 39.5 ± 9.5 | 38.8 ± 9.1 | 39.6 ± 9.7 | 40.4 ± 9.8 | n.s. |
| Disease duration (yrs), mean ± SD | 10.5 ± 7.6 | 10.5 ± 7.1 | 10.5 ± 8.2 | 10.2 ± 7.1 | n.s. |
| ARR in the 2 years prior FTY, mean ±SD | 0.82 ± 0.84 | 0.74 ± 0.83 | 0.93 ± 0.90 | 0.78 ± 0.73 | n.s. |
| Previous DMT | n.s. | ||||
| Naïve | 30 (7.9%) | 12 (7.9%) | 13 (8.6%) | 5 (6.5%) | |
| No therapy | 26 (6.8%) | 12 (7.9%) | 10 (6.6%) | 4 (5.2%) | |
| IFN | 149 (39.1%) | 59 (38.8%) | 58 (38.2%) | 32 (41.5%) | |
| GA | 104 (27.3%) | 40 (26.3%) | 40 (26.3%) | 24 (31.2%) | |
| DMF | 13 (3.4%) | 4 (2.6%) | 6 (3.9%) | 3 (3.9%) | |
| Teriflunomide | 11 (2.9%) | 4 (2.6%) | 3 (2%) | 4 (5.2%) | |
| Immunosuppressants | 17 (4.5%) | 6 (4.0%) | 9 (5.8%) | 2 (2.6%) | |
| Natalizumab | 29 (7.6%) | 13 (8.6%) | 13 (8.6%) | 3 (3.9%) | |
| Other | 2 (0.5%) | 2 (1.3%) | 0 (0%) | 0 (0%) | |
| EDSS at FTY start, median (range) | 2.0 (0–7.0) | 2 (0–6.0) | 2 (0–7.0) | 2 (0–6.0) | n.s. |
| <Patients with Gd+ lesions at baseline brain MRI scan | 33.5% | 35.1% | 31.7% | 33.8% | n.s. |
| Patients with new/enlarged T2 lesions at baseline brain MRI scan | 49.1% | 45.7% | 54.5% | 44.9% | n.s. |
| Model | Top-f | Min-fr | Sign | Ntree | Nodesize | Maxn | TR AUROC | TR AUPRC | TR F | TR Acc | TE AUROC | TE AUPRC | TE F | TE Acc |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g1 | 500 | 0.05 | 1022 | 20 | 10 | 10 | 0.8493 | 0.8476 | 0.7973 | 0.796 | 0.65 | 0.6483 | 0.6837 | 0.5194 |
| g2 | 500 | 0.1 | 123 | 10 | 10 | 30 | 0.8438 | 0.8496 | 0.7861 | 0.7565 | 0.6446 | 0.663 | 0.7142 | 0.5844 |
| g3 | 500 | 0.05 | 1022 | 10 | 1 | 15 | 0.838 | 0.8671 | 0.7567 | 0.7631 | 0.5801 | 0.5907 | 0.745 | 0.6623 |
| g4 | 500 | 0.15 | 8 | 10 | 2 | 60 | 0.858 | 0.8712 | 0.8 | 0.7828 | 0.6135 | 0.6176 | 0.7102 | 0.5974 |
| Description | Size | Expected | Enrichment | p Value | FDR |
|---|---|---|---|---|---|
| Renin secretion | 65 | 0.23 | 13.26 | 0.001 | 0.42 |
| Calcium signaling pathway | 183 | 0.64 | 6.28 | 0.003 | 0.51 |
| Sphingolipid signaling pathway | 118 | 0.41 | 7.30 | 0.008 | 0.65 |
| Sphingolipid metabolism | 47 | 0.16 | 12.22 | 0.011 | 0.65 |
| Cholesterol metabolism | 50 | 0.17 | 11.49 | 0.013 | 0.65 |
| Cell adhesion molecules (CAMs) | 144 | 0.50 | 5.98 | 0.013 | 0.65 |
| cGMP-PKG signaling pathway | 163 | 0.57 | 5.29 | 0.018 | 0.67 |
| Cortisol synthesis and secretion | 64 | 0.22 | 8.98 | 0.021 | 0.67 |
| Inflammatory bowel disease (IBD) | 65 | 0.23 | 8.84 | 0.021 | 0.67 |
| Long-term potentiation | 67 | 0.23 | 8.58 | 0.022 | 0.67 |
| Model | Top-f | Min-fr | Sign | Mtry | N Tree | Node Size | Maxn | TR AUROC | TR AUPRC | TR F | TR acc | TE AUROC | TE AUPRC | TE F | TE acc |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c1 | 8 | 0.5 | 9 | 10 | 100 | 2 | 100 | 1 | 1 | 1 | 1 | 0.6895 | 0.6709 | 0.7339 | 0.6494 |
| c2 | 8 | 0.5 | 9 | 4 | 20 | 2 | 100 | 0.9785 | 0.9803 | 0.9255 | 0.9211 | 0.6405 | 0.7320 | 0.7091 | 0.6364 |
| c3 | 8 | 0.5 | 9 | 10 | 20 | 2 | 30 | 0.9152 | 0.9281 | 0.8434 | 0.8289 | 0.623 | 0.6422 | 0.7379 | 0.6494 |
| c4 | 2 | 0.05 | 14 | 3 | 10 | 1 | 100 | 0.9971 | 0.9974 | 0.9684 | 1 | 0.6895 | 0.6709 | 0.7339 | 0.6494 |
| Model | TR_AUROC | TR_AUPRC | TR_F | TR_Acc | TE_AUROC | TE_AUPRC | TE_F | TE_acc |
|---|---|---|---|---|---|---|---|---|
| g2-c1 | 0.9997 | 0.9997 | 0.9933 | 0.9934 | 0.7095 | 0.7328 | 0.7328 | 0.6623 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrè, L.; Clarelli, F.; Pignolet, B.; Mascia, E.; Frasca, M.; Santoro, S.; Sorosina, M.; Bucciarelli, F.; Moiola, L.; Martinelli, V.; et al. Combining Clinical and Genetic Data to Predict Response to Fingolimod Treatment in Relapsing Remitting Multiple Sclerosis Patients: A Precision Medicine Approach. J. Pers. Med. 2023, 13, 122. https://doi.org/10.3390/jpm13010122
Ferrè L, Clarelli F, Pignolet B, Mascia E, Frasca M, Santoro S, Sorosina M, Bucciarelli F, Moiola L, Martinelli V, et al. Combining Clinical and Genetic Data to Predict Response to Fingolimod Treatment in Relapsing Remitting Multiple Sclerosis Patients: A Precision Medicine Approach. Journal of Personalized Medicine. 2023; 13(1):122. https://doi.org/10.3390/jpm13010122
Chicago/Turabian StyleFerrè, Laura, Ferdinando Clarelli, Beatrice Pignolet, Elisabetta Mascia, Marco Frasca, Silvia Santoro, Melissa Sorosina, Florence Bucciarelli, Lucia Moiola, Vittorio Martinelli, and et al. 2023. "Combining Clinical and Genetic Data to Predict Response to Fingolimod Treatment in Relapsing Remitting Multiple Sclerosis Patients: A Precision Medicine Approach" Journal of Personalized Medicine 13, no. 1: 122. https://doi.org/10.3390/jpm13010122
APA StyleFerrè, L., Clarelli, F., Pignolet, B., Mascia, E., Frasca, M., Santoro, S., Sorosina, M., Bucciarelli, F., Moiola, L., Martinelli, V., Comi, G., Liblau, R., Filippi, M., Valentini, G., & Esposito, F. (2023). Combining Clinical and Genetic Data to Predict Response to Fingolimod Treatment in Relapsing Remitting Multiple Sclerosis Patients: A Precision Medicine Approach. Journal of Personalized Medicine, 13(1), 122. https://doi.org/10.3390/jpm13010122