
Bioinformatic Analysis of Recurrent Genomic Alterations and Corresponding Pathway Alterations in Ewing Sarcoma


Abstract
:1. Introduction
2. Materials and Methods
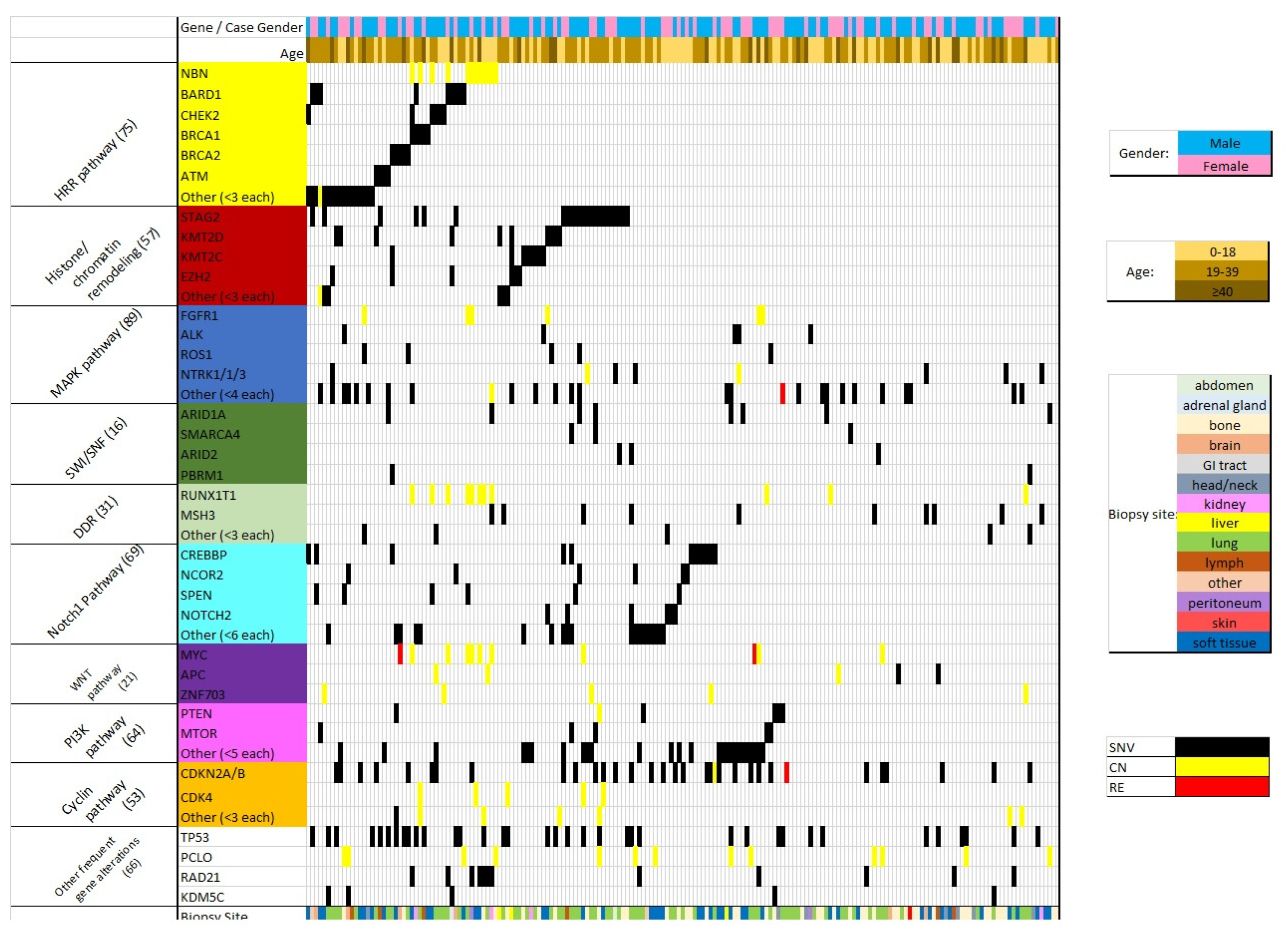
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Burningham, Z.; Hashibe, M.; Spector, L.; Schiffman, J.D. The epidemiology of sarcoma. Clin. Sarcoma Res. 2012, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.; Alava, E.; Kovar, H.; Sorenson, P.; Delattre, O.; Dirksen, U. Wing sarcoma. Nat. Rev. Dis. Primers 2018, 4, 5. [Google Scholar] [CrossRef]
- Womer, R.B.; West, D.C.; Krailo, M.D.; Dickman, P.; Pawel, B.; Grier, H.; Marcus, K.; Sailer, S.; Healey, J.; Dormans, J.; et al. Randomized Controlled Trial of Interval-Compressed Chemotherapy for the Treatment of Localized Ewing Sarcoma: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 4148–4154. [Google Scholar] [CrossRef]
- Barker, L.M.; Pendergrass, T.W.; Sanders, J.E.; Hawkins, D.S. Survival After Recurrence of Ewing’s Sarcoma Family of Tumors. J. Clin. Oncol. 2005, 23, 4354–4362. [Google Scholar] [CrossRef]
- Esiashvili, N.; Goodman, M.; Marcus, R.B. Changes in Incidence and Survival of Ewing Sarcoma Patients Over the Past 3 Decades: Surveillance Epidemiology and End Results Data. J. Pediatr. Hematol./Oncol. 2008, 30, 425–430. [Google Scholar] [CrossRef]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.; Ferrari, S.; Deley, M. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J. Clin. Oncol 2015, 33, 3036–3046. [Google Scholar] [CrossRef]
- The Ewing Family of Tumors—A Subgroup of Small-Round-Cell Tumors Defined by Specific Chimeric Transcripts|NEJM. Available online: https://www.nejm.org/doi/10.1056/NEJM199408043310503?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200www.ncbi.nlm.nih.gov (accessed on 29 June 2020).
- Denny, C.T. Special Article: Gene Rearrangements in Ewing’s Sarcoma. Cancer Investig. 2009, 14, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, P.H.B.; Lessnick, S.L.; Lopez-Terrada, D.; Liu, X.F.; Triche, T.J.; Denny, C.T. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS–family transcription factor, ERG. Nat. Genet. 1994, 6, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Jeon, I.S.; Davis, J.N.; Braun, B.S.; Sublett, J.; Roussel, M.; Denny, C.; Shapiro, D. A variant Ewing’s sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene 1995, 10, 1229–1234. [Google Scholar] [PubMed]
- Urano, F.; Umezawa, A.; Yabe, H.; Hong, W.; Yoshida, K.; Fujinaga, K.; Hata, J. Molecular analysis of Ewing’s sarcoma: Another fusion gene, EWS-E1AF, available for diagnosis. Jpn. J. Cancer Res. 1998, 89, 703–711. [Google Scholar] [CrossRef]
- Peter, M.; Couturier, J.; Pacquement, H.; Michon, J.; Thomas, G.; Magdelenat, H.; Delattre, O. A new member of the ETS family fused to EWS in Ewing tumors. Oncogene 1997, 14, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Minas, T.Z.; Surdez, D.; Javaheri, T.; Tanaka, M.; Howarth, M.; Kang, H.; Han, J.; Han, Z.; Sax, B.; Kream, B.; et al. Combined experience of six independent laboratories attempting to create an Ewing sarcoma mouse model. Oncotarget 2016, 8, 34141–34163. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.D.; Teitell, M.A.; Arvand, A.; Denny, C.T. Divergent Ewing’s sarcoma EWS/ETS fusions confer a common tumorigenic phenotype on NIH3T3 cells. Oncogene 1999, 18, 5506–5513. [Google Scholar] [CrossRef] [PubMed]
- Sole, A.; Grossetête, S.; Heintzé, M.; Babin, L.; Zaïdi, S.; Revy, P.; Renouf, B.; De Cian, A.; Giovannangeli, C.; Pierre-Eugène, C.; et al. Unraveling Ewing sarcoma tumorigenesis originating from patient-derived Mesenchymal Stem Cells. Cancer Res. 2021, 81, 4994–5006. [Google Scholar] [CrossRef] [PubMed]
- Cote, G.M.; He, J.; Choy, E. Next-Generation Sequencing for Patients with Sarcoma: A Single Center Experience. Oncologist 2018, 23, 234–242. [Google Scholar] [CrossRef]
- Brohl, A.S.; Solomon, D.A.; Chang, W.; Wang, J.; Song, Y.; Sindiri, S.; Patidar, R.; Hurd, L.; Chen, L.; Shern, J.F.; et al. The Genomic Landscape of the Ewing Sarcoma Family of Tumors Reveals Recurrent STAG2 Mutation. PLoS Genet. 2014, 10, e1004629. [Google Scholar] [CrossRef]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The Genomic Landscape of Pediatric Ewing Sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef]
- Sand, L.G.L.; Szuhai, K.; Hogendoorn, P.C.W. Sequencing Overview of Ewing Sarcoma: A Journey across Genomic, Epigenomic and Transcriptomic Landscapes. Int. J. Mol. Sci. 2015, 16, 16176–16215. [Google Scholar] [CrossRef]
- Tirode, F.; Surdez, D.; Ma, X.; Parker, M.; Deley, M.; Bahrami, A.; Zhang, Z.; Lapouble, E.; Grossetete-Lalami, S.; Rusch, M.; et al. Genomic Landscape of Ewing Sarcoma Defines an Aggressive Subtype with Co-Association of STAG2 and TP53 Mutations. Cancer Discov. 2014, 4, 1342–1353. [Google Scholar] [CrossRef]
- El Beaino, M.; Liu, J.; Wasylishen, A.R.; Pourebrahim, R.; Migut, A.; Bessellieu, B.J.; Huang, K.; Lin, P. Loss of Stag2 cooperates with EWS-FLI1 to transform murine Mesenchymal stem cells. BMC Cancer 2020, 20, 3. [Google Scholar] [CrossRef]
- Agelopoulos, K.; Richter, G.H.S.; Schmidt, E.; Dirksen, U.; von Heykin, K.; Moser, B.; Klein, H.; Kontny, U.; Dugas, M.; Poos, K.; et al. Deep Sequencing in Conjunction with Expression and Functional Analyses Reveals Activation of FGFR1 in Ewing Sarcoma. Clin. Cancer Res. 2015, 21, 4935–4946. [Google Scholar] [CrossRef]
- Mackintosh, C.; Ordóñez, J.L.; García-Domínguez, D.J.; Sevillano, V.; Llombart-Bosch, A.; Szuhai, K.; Scotlandi, K.; Alberghini, M.; Sciot, R.; Sinnaeve, F.; et al. 1q gain and CDT2 overexpression underlie an aggressive and highly proliferative form of Ewing sarcoma. Oncogene 2012, 31, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Sun, Y.; Lv, Z.; Zhang, Z.; Su, Q.; Wu, H.; Zhang, W.; Yuan, W.; Zuo, L.; Shi, L.; et al. Effects of FGFR4 G388R, V10I polymorphisms on the likelihood of cancer. Sci. Rep. 2021, 11, 1373. [Google Scholar] [CrossRef]
- Xu, W.; Li, Y.; Wang, X.; Chen, B.; Wang, Y.; Liu, S.; Xu, J.; Zhoa, W.; Wu, J. FGFR4 transmembrane domain polymorphism and cancer risk: A meta-analysis including 8555 subjects. Eur. J. Cancer 2010, 46, 3332–3338. [Google Scholar] [CrossRef]
- Xiong, S.W.; Ma, J.; Feng, F.; Fu, F.; Shu, S.-R.; Ma, T.; Wu, C.; Liu, G.-C.; Zhu, J. Functional FGFR4 Gly388Arg polymorphism contributes to cancer susceptibility: Evidence from meta-analysis. Oncotarget 2017, 8, 25300–25309. [Google Scholar] [CrossRef]
- Bange, J.; Prechtl, D.; Cheburkin, Y.; Specht, K.; Harbeck, N.; Schmitt, M.; Knyazeva, T.; Müller, S.; Gärtner, S.; Sures, I.; et al. Cancer Progression and Tumor Cell Motility Are Associated with the FGFR4 Arg388 Allele. Cancer Res. 2002, 62, 840–847. [Google Scholar]
- Seitzer, N.; Mayr, T.; Streit, S.; Ullrich, A. A Single Nucleotide Change in the Mouse Genome Accelerates Breast Cancer Progression. Cancer Res. 2010, 70, 802–812. [Google Scholar] [CrossRef]
- Ulaganathan, V.K.; Sperl, B.; Rapp, U.R.; Ullrich, A. Germline variant FGFR4 p.G388R exposes a membrane-proximal STAT3 binding site. Nature 2015, 528, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, Y.; Ozaki, T.; Ouchida, M.; Umehara, N.; Ohata, N.; Yoshida, A.; Shimizu, K.; Inoue, H. Single nucleotide polymorphism in fibroblast growth factor receptor 4 at codon 388 is associated with prognosis in high-grade soft tissue sarcoma. Cancer 2003, 98, 2245–2250. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Abdel-Wahab, O.; Nahas, M.K.; Wang, K.; Rampal, R.K.; Intelkofer, A.M.; Patel, J.; Kristov, A.; Framopton, G.M.; Young, L.E.; et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood 2016, 127, 3004–3014. [Google Scholar] [CrossRef]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyanm, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.; et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: The current landscape and barriers to clinical application. Oncotarget 2016, 8, 16052–16074. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.D.; Sarker, D.; Meyer, T.; Yau, T.; Macarulla, T.; Park, J.-W.; Choo, S.P.; Hollebecque, A.; Sung, M.W.; Lim, H.-Y.; et al. First-in-Human Phase I Study of Fisogatinib (BLU-554) Validates Aberrant FGF19 Signaling as a Driver Event in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.X.; Lamba, N.; Hwang, W.L.; Niemierko, A.; DuBois, S.G.; Haas-Kogan, D.A. Risk stratification by somatic mutation burden in Ewing sarcoma. Cancer 2019, 125, 1357–1364. [Google Scholar] [CrossRef]
- Adane, B.; Alexe, G.; Seong, B.K.A.; Lu, D.; Hwang, E.E.; Hnisz, D.; Lareau, C.A.; Ross, L.; Lin, S.; Dela Cruz, F.S.; et al. STAG2 loss rewires oncogenic and developmental programs to promote metastasis in Ewing sarcoma. Cancer Cell 2021, 39, 827–844.e10. [Google Scholar] [CrossRef] [PubMed]
- Surdez, D.; Zaidi, S.; Grossetête, S.; Laud-Duval, K.; Sole Ferre, A.; Mous, L.; Vourc’h, T.; Tirode, F.; Pierron, G.; Raynal, V.; et al. STAG2 mutations alter CTCF-anchored loop extrusion, reduce cis-regulatory interactions and EWSR1-FLI1 activity in Ewing sarcoma. Cancer Cell 2021, 39, 810–826.e9. [Google Scholar] [CrossRef] [PubMed]
- Witcher, M.; Emerson, B.M. Epigenetic silencing of the p16(INK4a) tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Mol. Cell 2009, 34, 271–284. [Google Scholar] [CrossRef]
- Benini, S.; Manara, M.C.; Cerisano, V.; Perdichizzi, S.; Strammiello, R.; Serra, M.; Picci, P.; Scotlandi, K. Contribution of MEK/MAPK and PI3-K signaling pathway to the malignant behavior of Ewing’s sarcoma cells: Therapeutic prospects. Int. J. Cancer 2004, 108, 358–366. [Google Scholar] [CrossRef]
- Niemeyer, B.F.; Parrish, J.K.; Spoelstra, N.S.; Joyal, T.; Richer, J.K.; Jedlicka, P. Variable Expression of PIK3R3 and PTEN in Ewing Sarcoma Impacts Oncogenic Phenotypes. PLoS ONE 2015, 10, e0120830. [Google Scholar] [CrossRef]
- Scotlandi, K.; Benini, S.; Nanni, P.; Lollini, P.L.; Nicoletti, G.; Landuzzi, L.; Serra, M.; Manara, M.C.; Picci, P.; Baldini, N. Blockage of Insulin-like Growth Factor-I Receptor Inhibits the Growth of Ewing’s Sarcoma in Athymic Mice. Cancer Res. 1998, 58, 4127–4131. [Google Scholar]
- Yamamoto, T.; Ohno, T.; Wakahara, K.; Nagano, A.; Kawai, G.; Saitou, M.; Takigami, I.; Matsuhashi, A.; Yamada, K.; Shimizu, K. Simultaneous inhibition of mitogen-activated protein kinase and phosphatidylinositol 3-kinase pathways augment the sensitivity to actinomycin D in Ewing sarcoma. J. Cancer Res. Clin. Oncol. 2009, 135, 1125–1136. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Meurette, O.; Mehlen, P. Notch Signaling in the Tumor Microenvironment. Cancer Cell 2018, 34, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Azaro, A.; Soria, J.C.; Lassen, U.; Le Tourneau, C.; Sarker, D.; Smith, C.; Ohnmacht, U.; Oakley, G.; Patel, B.K.R.; et al. First-in-human study of LY3039478, an oral Notch signaling inhibitor in advanced or metastatic cancer. Ann. Oncol. 2018, 29, 1911–1917. [Google Scholar] [CrossRef]
- Lee, J.-M.; Ledermann, J.A.; Kohn, E.C. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann. Oncol. 2014, 25, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Applying Synthetic Lethality for the Selective Targeting of Cancer|NEJM. Available online: https://www.nejm.org/doi/10.1056/NEJMra1407390?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed (accessed on 11 January 2021).
- EWS–FLI1 Increases Transcription to Cause R-Loops and Block BRCA1 Repair in Ewing Sarcoma. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6318124/ (accessed on 1 December 2020).
- Brenner, J.C.; Feng, F.Y.; Han, S.; Patel, S.; Goyal, S.V.; Bou-Marou, L.M.; Liu, M.; Lonigro, R.; Presner, J.R.; Tomlins, S.A.; et al. PARP-1 Inhibition as a Targeted Strategy to Treat Ewing’s Sarcoma. Cancer Res. 2012, 72, 1608–1613. [Google Scholar] [CrossRef]
- Stewart, E.; Goshorn, R.; Bradley, C.; Griffiths, L.M.; Benavente, C.; Twarog, N.R.; Miller, G.M.; Caufield, W.; Freeman, B.B.; Bahrami, A.; et al. Targeting the DNA Repair Pathway in Ewing Sarcoma. Cell Rep. 2014, 9, 829–841. [Google Scholar] [CrossRef]
- Chugh, R.; Ballman, K.V.; Helman, L.J.; Patel, S.; Whelan, J.S.; Widemann, B.; Lu, Y.; Hawkins, D.S.; Mascarenhas, L.; Glod, J.W.; et al. SARC025 arms 1 and 2: A phase 1 study of the poly(ADP-ribose) polymerase inhibitor niraparib with temozolomide or irinotecan in patients with advanced Ewing sarcoma. Cancer 2021, 127, 1301–1310. [Google Scholar] [CrossRef]
- Haugen, A.C.; Goel, A.; Yamada, K.; Marra, G.; Nguyen, T.P.; Nagasaka, T.; Kanazawa, S.; Koike, J.; Kikuchi, Y.; Zhong, X.; et al. Genetic Instability Caused by Loss of MutS Homologue 3 in Human Colorectal Cancer. Cancer Res. 2008, 68, 8465–8472. [Google Scholar] [CrossRef]
- Davis, J.N.; McGhee, L.; Meyers, S. The ETO (MTG8) gene family. Gene 2003, 303, 1–10. [Google Scholar] [CrossRef]
- Linqing, Z.; Guohua, J.; Haoming, L.; Xuelei, T.; Jianbing, Q.; Meiling, T. Runx1t1 Regulates the Neuronal Differentiation of Radial Glial Cells from the Rat Hippocampus. Stem. Cells Transl. Med. 2015, 4, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.H.; Chang, S.J.; Chang, H.C.; Chien, C.L.; Huang, T.S.; Feng, T.C.; Lin, W.W.; Shih, C.C.; Yang, M.H.; Yang, S.H.; et al. Endothelial angiogenesis is directed by RUNX1T1-regulated VEGFA, BMP4 and TGF-β2 expression. PLoS ONE 2017, 12, e0179758. [Google Scholar] [CrossRef] [PubMed]
- Baby, N.; Li, Y.; Ling, E.A.; Lu, J.; Dheen, S.T. Runx1t1 (Runt-Related Transcription Factor 1; Translocated to, 1) Epigenetically Regulates the Proliferation and Nitric Oxide Production of Microglia. PLoS ONE 2014, 9, e89326. [Google Scholar] [CrossRef] [PubMed]
- Phelan, M.; Sif, S.; Narlikar, G.; Kingston, R. Reconstitution of a Core Chromatin Remodeling Complex from SWI/SNF Subunits. Mol. Cell 1999, 3, 247–253. [Google Scholar] [CrossRef]
- Wu, J.N.; Roberts, C.W.M. ARID1A Mutations in Cancer: Another Epigenetic Tumor Suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef]
- Mathur, R.; Alver, B.H.; San Roman, A.K.; Wilson, B.G.; Wang, X.; Agoston, A.T.; Park, P.J.; Shivdasani, R.A.; Roberts, C.W.M. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 2017, 49, 296–302. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Gounder, M.; Schöffski, P.; Jones, R.L.; Agulnik, M.; Cote, G.M.; Villalobos, V.M.; Attia, S.; Chugh, R.; Chen, T.W.W.; Jahan, T.; et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: An international, open-label, phase 2 basket study. Lancet Oncol. 2020, 21, 1423–1432. [Google Scholar] [CrossRef]
- Bonasio, R.; Tu, S.; Reinberg, D. Molecular Signals of Epigenetic States. Science 2010, 330, 612–616. [Google Scholar] [CrossRef]
- Bennani-Baiti, I.M.; Machado, I.; Llombart-Bosch, A.; Kovar, H. Lysine-specific demethylase 1 (LSD1/KDM1A/AOF2/BHC110) is expressed and is an epigenetic drug target in chondrosarcoma, Ewing’s sarcoma, osteosarcoma, and rhabdomyosarcoma. Hum. Pathol. 2012, 43, 1300–1307. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Vallurupalli, M.; Chen, L.; Crompton, B.; Cowley, G.; Vazquez, F.; Weir, B.A.; Tsherniak, A.; Parasuraman, S.; Kim, S.; et al. Functional, chemical genomic, and super-enhancer screening identify sensitivity to cyclin D1/CDK4 pathway inhibition in Ewing sarcoma. Oncotarget 2015, 6, 30178–30193. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef] [PubMed]
- Zarkowska, T.; Mittnacht, S. Differential Phosphorylation of the Retinoblastoma Protein by G1/S Cyclin-dependent Kinases. J. Biol. Chem. 1997, 272, 12738–12746. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, D.; de Hooge, A.S.; Santos, S.J.; Horst, D.; Wiertz, E.J.; van Eggermond, M.C.; van den Elsen, P.J.; Taminiau, A.H.M.; Ottaviano, L.; Schaefer, K.L.; et al. Reduced human leukocyte antigen expression in advanced-stage Ewing sarcoma: Implications for immune recognition. J. Pathol. 2009, 218, 222–231. [Google Scholar] [CrossRef]
- Vargas, A.C.; Maclean, F.M.; Sioson, L.; Tran, D.; Bonar, F.; Mahar, A.; Cheah, A.L.; Russell, P.; Grimson, P.; Richardson, L.; et al. Prevalence of PD-L1 expression in matched recurrent and/or metastatic sarcoma samples and in a range of selected sarcomas subtypes. PLoS ONE 2020, 15, e0222551. [Google Scholar] [CrossRef] [PubMed]
- Alldinger, I.; Schaefer, K.L.; Goedde, D.; Ottaviano, L.; Dirksen, U.; Ranft, A.; Juergens, H.; Gabbert, H.E.; Knoefel, W.T.; Poremba, C. Microsatellite instability in Ewing tumor is not associated with loss of mismatch repair protein expression. J. Cancer Res. Clin. Oncol. 2007, 133, 749–759. [Google Scholar] [CrossRef]
- Ebinger, M.; Bock, T.; Kandolf, R.; Sotlar, K.; Bültmann, B.D.; Greil, J. Standard mono- and dinucleotide repeats do not appear to be sensitive markers of microsatellite instability in the Ewing family of tumors. Cancer Genet. Cytogenet. 2005, 157, 189–190. [Google Scholar] [CrossRef]
- Tarkkanen, M.; Aaltonen, L.A.; Böhling, T.; Kivioja, A.; Karaharju, E.; Elomaa, I.; Knuutila, S. No evidence of microsatellite instability in bone tumours. Br. J. Cancer. 1996, 74, 453–455. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Total | Percent | |
|---|---|---|
| Gender | ||
| Male | 113 | 60% |
| Female | 76 | 40% |
| Age | ||
| 0–18 | 75 | 40% |
| 19–39 | 87 | 46% |
| ≥40 | 27 | 14% |
| Genomic Alteration | No FGFR4 G388R Variant | FGFR4 G388R Variant Present | Percentage of Samples with Pathway Alteration | Percentage of Total Samples with GA and FGFR G388R Variant |
|---|---|---|---|---|
| Total | 92 | 97 | 51 | 43% |
| MAPK | 26 | 63 | 25 | 33% |
| NOTCH1 | 32 | 37 | 23 | 20% |
| HRR | 39 | 36 | 24 | 19% |
| Histone/Chromatin Remodeling | 24 | 33 | 24 | 18% |
| Cyclin | 25 | 28 | 23 | 15% |
| PI3K | 45 | 19 | 20 | 9% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rock, A.; Uche, A.; Yoon, J.; Agulnik, M.; Chow, W.; Millis, S. Bioinformatic Analysis of Recurrent Genomic Alterations and Corresponding Pathway Alterations in Ewing Sarcoma. J. Pers. Med. 2023, 13, 1499. https://doi.org/10.3390/jpm13101499
Rock A, Uche A, Yoon J, Agulnik M, Chow W, Millis S. Bioinformatic Analysis of Recurrent Genomic Alterations and Corresponding Pathway Alterations in Ewing Sarcoma. Journal of Personalized Medicine. 2023; 13(10):1499. https://doi.org/10.3390/jpm13101499
Chicago/Turabian StyleRock, Adam, An Uche, Janet Yoon, Mark Agulnik, Warren Chow, and Sherri Millis. 2023. "Bioinformatic Analysis of Recurrent Genomic Alterations and Corresponding Pathway Alterations in Ewing Sarcoma" Journal of Personalized Medicine 13, no. 10: 1499. https://doi.org/10.3390/jpm13101499
APA StyleRock, A., Uche, A., Yoon, J., Agulnik, M., Chow, W., & Millis, S. (2023). Bioinformatic Analysis of Recurrent Genomic Alterations and Corresponding Pathway Alterations in Ewing Sarcoma. Journal of Personalized Medicine, 13(10), 1499. https://doi.org/10.3390/jpm13101499