
The Role of Uric Acid in Human Health: Insights from the Uricase Gene


Abstract
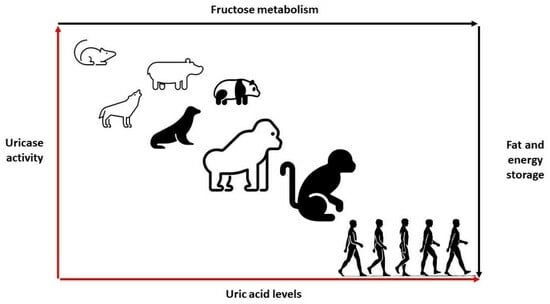
:
1. Background
2. Genetic Correlation between Urate Levels and Cardiometabolic Traits
3. Uricase Activity: Fructose Metabolism and Energy Storage
4. Regulating Blood Pressure: The Role of Salt and Uric Acid
5. Hyperuricemia and Hypertension: A Cause or Effect?
6. Uric Acid and Neurodegenerative Diseases: The Antioxidant Hypothesis
7. Hyperuricemia and Innate Immune System: Acquired Protection?
8. Uric Acid: A Therapeutic Target or Disease Bystander?
9. Current and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, R.J.; Titte, S.; Cade, J.R.; Rideout, B.A.; Oliver, W.J. Uric acid, evolution and primitive cultures. Semin. Nephrol. 2005, 25, 3–8. [Google Scholar] [CrossRef]
- Alvarez-Lario, B.; Macarron-Vicente, J. Uric acid and evolution. Rheumatology 2010, 49, 2010–2015. [Google Scholar] [CrossRef]
- Roman, Y.M. The Daniel K. Inouye College of Pharmacy Scripts: Perspectives on the Epidemiology of Gout and Hyperuricemia. Hawaii J. Med. Public Health 2019, 78, 71–76. [Google Scholar]
- Liu, Y.; Jarman, J.B.; Low, Y.S.; Augustijn, H.E.; Huang, S.; Chen, H.; DeFeo, M.E.; Sekiba, K.; Hou, B.-H.; Meng, X.; et al. A widely distributed gene cluster compensates for uricase loss in hominids. Cell 2023, 186, 3400–3413.e20. [Google Scholar] [CrossRef]
- Tong, Y.; Wei, Y.; Ju, Y.; Li, P.; Zhang, Y.; Li, L.; Gao, L.; Liu, S.; Liu, D.; Hu, Y.; et al. Anaerobic purinolytic enzymes enable dietary purine clearance by engineered gut bacteria. Cell Chem. Biol. 2023. [Google Scholar] [CrossRef]
- Wei, J.; Zhang, Y.; Dalbeth, N.; Terkeltaub, R.; Yang, T.; Wang, Y.; Yang, Z.; Li, J.; Wu, Z.; Zeng, C.; et al. Association Between Gut Microbiota and Elevated Serum Urate in Two Independent Cohorts. Arthritis Rheumatol. 2022, 74, 682–691. [Google Scholar] [CrossRef]
- Kim, H.W.; Yoon, E.-J.; Jeong, S.H.; Park, M.-C. Distinct Gut Microbiota in Patients with Asymptomatic Hyperuricemia: A Potential Protector against Gout Development. Yonsei Med. J. 2022, 63, 241–251. [Google Scholar] [CrossRef]
- Mouradjian, M.T.; Plazak, M.E.; Gale, S.E.; Noel, Z.R.; Watson, K.; Devabhakthuni, S. Pharmacologic Management of Gout in Patients with Cardiovascular Disease and Heart Failure. Am. J. Cardiovasc. Drugs 2020, 20, 431–445. [Google Scholar] [CrossRef]
- Becker, M.A.; Baraf, H.S.B.; Yood, R.A.; Dillon, A.; Vazquez-Mellado, J.; Ottery, F.D.; Khanna, D.; Sundy, J.S. Long-term safety of pegloticase in chronic gout refractory to conventional treatment. Ann. Rheum. Dis. 2013, 72, 1469–1474. [Google Scholar] [CrossRef]
- Sundy, J.S.; Baraf, H.S.B.; Yood, R.A.; Edwards, N.L.; Gutierrez-Urena, S.R.; Treadwell, E.L.; Vazquez-Mellado, J.; White, W.B.; Lipsky, P.E.; Horowitz, Z.; et al. Efficacy and tolerability of pegloticase for the treatment of chronic gout in patients refractory to conventional treatment: Two randomized controlled trials. JAMA 2011, 306, 711–720. [Google Scholar] [CrossRef]
- Hershfield, M.S.; Roberts, L.J., 2nd; Ganson, N.J.; Kelly, S.J.; Santisteban, I.; Scarlett, E.; Jaggers, D.; Sundy, J.S. Treating gout with pegloticase, a PEGylated urate oxidase, provides insight into the importance of uric acid as an antioxidant in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 14351–14356. [Google Scholar] [CrossRef]
- Yang, X.; Yuan, Y.; Zhan, C.-G.; Liao, F. Uricases as therapeutic agents to treat refractory gout: Current states and future directions. Drug Dev. Res. 2012, 73, 66–72. [Google Scholar] [CrossRef]
- Watanabe, S.; Kang, D.-H.; Feng, L.; Nakagawa, T.; Kanellis, J.; Lan, H.; Mazzali, M.; Johnson, R.J. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension 2002, 40, 355–360. [Google Scholar] [CrossRef]
- Johnson, R.J.; Stenvinkel, P.; Andrews, P.; Sanchez-Lozada, L.G.; Nakagawa, T.; Gaucher, E.; Andres-Hernando, A.; Rodriguez-Iturbe, B.; Jimenez, C.R.; Garcia, G.; et al. Fructose metabolism as a common evolutionary pathway of survival associated with climate change, food shortage and droughts. J. Intern. Med. 2020, 287, 252–262. [Google Scholar] [CrossRef]
- Kratzer, J.T.; Lanaspa, M.A.; Murphy, M.N.; Cicerchi, C.; Graves, C.L.; Tipton, P.A.; Ortlund, E.A.; Johnson, R.J.; Gaucher, E.A. Evolutionary history and metabolic insights of ancient mammalian uricases. Proc. Natl. Acad. Sci. USA 2014, 111, 3763–3768. [Google Scholar] [CrossRef]
- Wu, X.; Muzny, D.M.; Lee, C.C.; Caskey, C.T. Two independent mutational events in the loss of urate oxidase during hominoid evolution. J. Mol. Evol. 1992, 34, 78–84. [Google Scholar] [CrossRef]
- Li, Z.; Hoshino, Y.; Tran, L.; Gaucher, E.A. Phylogenetic Articulation of Uric Acid Evolution in Mammals and How It Informs a Therapeutic Uricase. Mol. Biol. Evol. 2022, 39, msab312. [Google Scholar] [CrossRef]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef]
- Drouin, G.; Godin, J.-R.; Page, B. The genetics of vitamin C loss in vertebrates. Curr. Genom. 2011, 12, 371–378. [Google Scholar] [CrossRef]
- Roman, Y.M. Moving the Needle in Gout Management: The Role of Culture, Diet, Genetics, and Personalized Patient Care Practices. Nutrients 2022, 14, 3590. [Google Scholar] [CrossRef]
- San Gabriel, D.E.D.; Slark, J. The association of gout with an increased risk of hypertension and diabetes mellitus among stroke survivors in New Zealand: A cross-sectional study using routinely collected electronic health data. JRSM Cardiovasc. Dis. 2019, 8, 2048004019863239. [Google Scholar] [CrossRef] [PubMed]
- Klemp, P.; Stansfield, S.A.; Castle, B.; Robertson, M.C. Gout is on the increase in New Zealand. Ann. Rheum. Dis. 1997, 56, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Kolz, M.; Johnson, T.; Sanna, S.; Teumer, A.; Vitart, V.; Perola, M.; Mangino, M.; Albrecht, E.; Wallace, C.; Farrall, M.; et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 2009, 5, e1000504. [Google Scholar] [CrossRef] [PubMed]
- Kottgen, A.; Albrecht, E.; Teumer, A.; Vitart, V.; Krumsiek, J.; Hundertmark, C.; Pistis, G.; Ruggiero, D.; O’Seaghdha, C.M.; Haller, T.; et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet. 2013, 45, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Alghubayshi, A.; Edelman, A.; Alrajeh, K.; Roman, Y. Genetic assessment of hyperuricemia and gout in Asian, Native Hawaiian, and Pacific Islander subgroups of pregnant women: Biospecimens repository cross-sectional study. BMC Rheumatol. 2022, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Butler, F.; Alghubayshi, A.; Roman, Y. The Epidemiology and Genetics of Hyperuricemia and Gout across Major Racial Groups: A Literature Review and Population Genetics Secondary Database Analysis. J. Pers. Med. 2021, 11, 231. [Google Scholar] [CrossRef]
- Phipps-Green, A.J.; Hollis-Moffatt, J.E.; Dalbeth, N.; Merriman, M.E.; Topless, R.; Gow, P.J.; Harrison, A.A.; Highton, J.; Jones, P.B.; Stamp, L.K.; et al. A strong role for the ABCG2 gene in susceptibility to gout in New Zealand Pacific Island and Caucasian, but not Māori, case and control sample sets. Hum. Mol. Genet. 2010, 19, 4813–4819. [Google Scholar] [CrossRef]
- Tin, A.; German Chronic Kidney Disease Study; Marten, J.; Halperin Kuhns, V.L.; Li, Y.; Wuttke, M.; Kirsten, H.; Sieber, K.B.; Qiu, C.; Gorski, M.; et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat. Genet. 2019, 51, 1459–1474. [Google Scholar] [CrossRef]
- Dong, Z.; Zhou, J.; Jiang, S.; Li, Y.; Zhao, D.; Yang, C.; Ma, Y.; Wang, Y.; He, H.; Ji, H.; et al. Effects of multiple genetic loci on the pathogenesis from serum urate to gout. Sci. Rep. 2017, 7, 43614. [Google Scholar] [CrossRef]
- Reynolds, R.J.; Irvin, M.R.; Bridges, S.L.; Kim, H.; Merriman, T.R.; Arnett, D.K.; Singh, J.A.; Sumpter, N.A.; Lupi, A.S.; Vazquez, A.I. Genetic correlations between traits associated with hyperuricemia, gout, and comorbidities. Eur. J. Hum. Genet. 2021, 29, 1438–1445. [Google Scholar] [CrossRef]
- Singh, J.A.; Cleveland, J.D. Gout and the risk of incident atrial fibrillation in older adults: A study of US Medicare data. RMD Open 2018, 4, e000712. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.P.; Pandya, B.J.; Choi, H.K. Comorbidities of Gout and Hyperuricemia in the US General Population: NHANES 2007-2008. Am. J. Med. 2012, 125, 679–687. [Google Scholar] [CrossRef]
- Singh, J.A.; Cleveland, J.D. Gout is associated with a higher risk of chronic renal disease in older adults: A retrospective cohort study of U.S. Medicare population. BMC Nephrol. 2019, 20, 93. [Google Scholar] [CrossRef]
- Zhu, Y.; Pandya, B.J.; Choi, H.K. Prevalence of gout and hyperuricemia in the US general population: The National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011, 63, 3136–3141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Wang, Y.; Fu, T.; Zhou, W.; Ge, X.; Sha, X.; Guo, J.; Dong, C.; Guo, G. Gout and risk of diabetes mellitus: Meta-analysis of observational studies. Psychol. Health Med. 2020, 25, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Lyngdoh, T.; Vuistiner, P.; Marques-Vidal, P.; Rousson, V.; Waeber, G.; Vollenweider, P.; Bochud, M. Serum uric acid and adiposity: Deciphering causality using a bidirectional Mendelian randomization approach. PLoS ONE 2012, 7, e39321. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Cicerchi, C.; Li, N.; Roncal-Jimenez, C.A.; Ishimoto, T.; Le, M.; Garcia, G.E.; Thomas, J.B.; Rivard, C.J.; et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS ONE 2012, 7, e47948. [Google Scholar] [CrossRef]
- Song, Z.; Roncal-Jimenez, C.A.; Lanaspa-Garcia, M.A.; Oppelt, S.A.; Kuwabara, M.; Jensen, T.; Milagres, T.; Andres-Hernando, A.; Ishimoto, T.; Garcia, G.E.; et al. Role of fructose and fructokinase in acute dehydration-induced vasopressin gene expression and secretion in mice. J. Neurophysiol. 2017, 117, 646–654. [Google Scholar] [CrossRef]
- Andres-Hernando, A.; Jensen, T.J.; Kuwabara, M.; Orlicky, D.J.; Cicerchi, C.; Li, N.; Roncal-Jimenez, C.A.; Garcia, G.E.; Ishimoto, T.; Maclean, P.S.; et al. Vasopressin mediates fructose-induced metabolic syndrome by activating the V1b receptor. JCI Insight 2021, 6, e140848. [Google Scholar] [CrossRef]
- Johnson, R.J.; Perez-Pozo, S.E.; Sautin, Y.Y.; Manitius, J.; Sanchez-Lozada, L.G.; Feig, D.I.; Shafiu, M.; Segal, M.; Glassock, R.J.; Shimada, M.; et al. Hypothesis: Could excessive fructose intake and uric acid cause type 2 diabetes? Endocr. Rev. 2009, 30, 96–116. [Google Scholar] [CrossRef]
- Johnson, R.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Shafiu, M.; Sundaram, S.; Le, M.; Ishimoto, T.; Sautin, Y.Y.; Lanaspa, M.A. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 2013, 62, 3307–3315. [Google Scholar] [CrossRef]
- Andres-Hernando, A.; Orlicky, D.J.; Kuwabara, M.; Ishimoto, T.; Nakagawa, T.; Johnson, R.J.; Lanaspa, M.A. Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab. 2020, 32, 117–127.e3. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Sanchez-Lozada, L.G.; Nakagawa, T.; Rodriguez-Iturbe, B.; Tolan, D.; Gaucher, E.A.; Andrews, P.; Lanaspa, M.A. Do thrifty genes exist? Revisiting uricase. Obesity 2022, 30, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Grillo, A.; Salvi, L.; Coruzzi, P.; Salvi, P.; Parati, G. Sodium Intake and Hypertension. Nutrients 2019, 11, 1970. [Google Scholar] [CrossRef] [PubMed]
- Mazzali, M.; Kanellis, J.; Han, L.; Feng, L.; Xia, Y.-Y.; Chen, Q.; Kang, D.-H.; Gordon, K.L.; Watanabe, S.; Nakagawa, T.; et al. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am. J. Physiol. Renal. Physiol. 2002, 282, F991–F997. [Google Scholar] [CrossRef]
- Mazzali, M.; Hughes, J.; Kim, Y.-G.; Jefferson, J.A.; Kang, D.-H.; Gordon, K.L.; Lan, H.Y.; Kivlighn, S.; Johnson, R.J. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001, 38, 1101–1106. [Google Scholar] [CrossRef]
- Andrés, M. Gout and Cardiovascular Disease: Mechanisms, Risk Estimations, and the Impact of Therapies. Gout Urate Cryst. Depos. Dis. 2023, 1, 152–166. [Google Scholar] [CrossRef]
- Klauser, A.S.; Halpern, E.J.; Strobl, S.; Gruber, J.; Feuchtner, G.; Bellmann-Weiler, R.; Weiss, G.; Stofferin, H.; Jaschke, W. Dual-Energy Computed Tomography Detection of Cardiovascular Monosodium Urate Deposits in Patients with Gout. JAMA Cardiol. 2019, 4, 1019–1028. [Google Scholar] [CrossRef]
- Wang, X.; Liu, X.; Qi, Y.; Zhang, S.; Shi, K.; Lin, H.; Grossfeld, P.; Wang, W.; Wu, T.; Qu, X.; et al. High Level of Serum Uric Acid induced Monocyte Inflammation is Related to Coronary Calcium Deposition in the Middle-Aged and Elder Population of China: A five-year Prospective Cohort Study. J. Inflamm. Res. 2022, 15, 1859–1872. [Google Scholar] [CrossRef]
- Mills, K.T.; Stefanescu, A.; He, J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237. [Google Scholar] [CrossRef]
- Kuwabara, M.; Niwa, K.; Nishi, Y.; Mizuno, A.; Asano, T.; Masuda, K.; Komatsu, I.; Yamazoe, M.; Takahashi, O.; Hisatome, I. Relationship between serum uric acid levels and hypertension among Japanese individuals not treated for hyperuricemia and hypertension. Hypertens. Res. 2014, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, Y.; Asayama, K.; Morimoto, A.; Satoh, M.; Sonoda, N.; Miyamatsu, N.; Ohno, Y.; Miyamoto, Y.; Izawa, S.; Ohkubo, T. Hyperuricemia predicts the risk for developing hypertension independent of alcohol drinking status in men and women: The Saku study. Hypertens. Res. 2020, 43, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bao, Y.; Meng, M.; Yu, L.; Lu, Y.; Sa, R.; Liang, X.; Shi, J. Association of serum uric acid with risk of stroke in US adults: A cross-sectional study from NHANES 1999–2020. J. Stroke Cerebrovasc. Dis. 2023, 32, 107206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, Y.; Wang, K.; Yin, R.; Pan, X.; Ma, A. Association between uric acid and the prognosis of acute ischemic stroke: A systematic review and meta-analysis. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 3016–3023. [Google Scholar] [CrossRef]
- Nakamura, K.; Ueki, K.; Matsuo, R.; Kiyohara, T.; Irie, F.; Wakisaka, Y.; Ago, T.; Kamouchi, M.; Kitazono, T.; Fukuoka Stroke Registry, I. Association between decreases in serum uric acid levels and unfavorable outcomes after ischemic stroke: A multicenter hospital-based observational study. PLoS ONE 2023, 18, e0287721. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Nie, X.; Leng, X.; Wang, D.; Pan, Y.; Yan, H.; Yang, Z.; Wen, M.; Pu, Y.; Zhang, Z.; et al. Increased serum uric acid level is associated with better outcome after endovascular treatment for acute ischemic stroke—A prospective cohort study. Ann. Transl. Med. 2022, 10, 1111. [Google Scholar] [CrossRef] [PubMed]
- Vo, V.; Lopez, G.; Malay, S.; Roman, Y.M. Cardiovascular Risk Factors Among Asian Americans: Perspectives on the Role of Acculturation in Cardiovascular Diseases Health Disparities. J. Immigr. Minor. Health 2023. [Google Scholar] [CrossRef]
- Coronado, G.; Chio-Lauri, J.; Cruz, R.D.; Roman, Y.M. Health Disparities of Cardiometabolic Disorders Among Filipino Americans: Implications for Health Equity and Community-Based Genetic Research. J. Racial Ethn. Health Disparities 2022, 9, 2560–2567. [Google Scholar] [CrossRef]
- Yano, K.; Rhoads, G.; Kagan, A. Epidemiology of serum uric acid among 8000 Japanese-American men in Hawaii. J. Chronic. Dis. 1977, 30, 171–184. [Google Scholar] [CrossRef]
- Shai, A.; Rimar, D.; Rozenbaum, M.; Wolfovitz, E.; Rosner, I. Gout in young migrant Filipino women in Israel: A changing epidemiology. Case reports and review of the literature. Rheumatol. Int. 2010, 30, 1685–1687. [Google Scholar] [CrossRef]
- Roman, Y.M.; McClish, D.; Price, E.T.; Sabo, R.T.; Woodward, O.M.; Mersha, T.B.; Shah, N.; Armada, A.; Terkeltaub, R. Cardiometabolic genomics and pharmacogenomics investigations in Filipino Americans: Steps towards precision health and reducing health disparities. Am. Heart J. Plus Cardiol. Res. Pract. 2022, 15, 100136. [Google Scholar] [CrossRef] [PubMed]
- Healey, L.A.; Bayani-Sioson, P.S. A defect in the renal excretion of uric acid in Filipinos. Arthritis Rheum. 1971, 14, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Montoye, H.J.; Mikkelsen, W.M. Serum uric acid and achievement in high school. Arthritis Rheum. 1973, 16, 359–362. [Google Scholar] [CrossRef]
- Inouye, E.; Park, K.S.; Asaka, A. Blood uric acid level and IQ: A study in twin families. Acta Genet. Med. Gemellol. Twin Res. 1984, 33, 237–242. [Google Scholar] [CrossRef]
- Kennett, K.F.; Cropley, A.J. Uric acid and divergent thinking: A possible relationship. Br. J. Psychol. 1975, 66, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Dubreuil, M.; Zhang, Y.; Neogi, T.; Rai, S.K.; Ascherio, A.; Hernan, M.A.; Choi, H.K. Gout and the risk of Alzheimer’s disease: A population-based, BMI-matched cohort study. Ann. Rheum. Dis. 2016, 75, 547–551. [Google Scholar] [CrossRef]
- Singh, J.A.; Cleveland, J.D. Gout and dementia in the elderly: A cohort study of Medicare claims. BMC Geriatr. 2018, 18, 281. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhong, S.; Liang, Y.; Zhang, X.; Zhang, R.; Kang, K.; Qu, H.; Xu, Y.; Zhao, C.; Zhao, M. Serum Uric Acid and the Risk of Dementia: A Systematic Review and Meta-Analysis. Front. Aging Neurosci. 2021, 13, 625690. [Google Scholar] [CrossRef]
- McFarland, N.R.; Burdett, T.; Desjardins, C.A.; Frosch, M.P.; Schwarzschild, M.A. Postmortem brain levels of urate and precursors in Parkinson’s disease and related disorders. Neurodegener. Dis. 2013, 12, 189–198. [Google Scholar] [CrossRef]
- Fazlollahi, A.; Zahmatyar, M.; Alizadeh, H.; Noori, M.; Jafari, N.; Nejadghaderi, S.A.; Sullman, M.J.M.; Gharagozli, K.; Kolahi, A.-A.; Safiri, S. Association between gout and the development of Parkinson’s disease: A systematic review and meta-analysis. BMC Neurol. 2022, 22, 383. [Google Scholar] [CrossRef]
- Tana, C.; Ticinesi, A.; Prati, B.; Nouvenne, A.; Meschi, T. Uric Acid and Cognitive Function in Older Individuals. Nutrients 2018, 10, 975. [Google Scholar] [CrossRef] [PubMed]
- Seifar, F.; Dinasarapu, A.R.; Jinnah, H.A. Uric Acid in Parkinson’s Disease: What Is the Connection? Mov. Disord. 2022, 37, 2173–2183. [Google Scholar] [CrossRef] [PubMed]
- Roman, Y. Pathway for ascertaining the role of uric acid in neurodegenerative diseases. Alzheimers Dement. 2022, 14, e12329. [Google Scholar] [CrossRef]
- Gersch, C.; Palii, S.P.; Imaram, W.; Kim, K.M.; Karumanchi, S.A.; Angerhofer, A.; Johnson, R.J.; Henderson, G.N. Reactions of peroxynitrite with uric acid: Formation of reactive intermediates, alkylated products and triuret, and in vivo production of triuret under conditions of oxidative stress. Nucleosides Nucleotides Nucleic Acids 2009, 28, 118–149. [Google Scholar] [CrossRef] [PubMed]
- Kanabrocki, E.L.; Ryan, M.D.; Hermida, R.C.; Ayala, D.E.; Scott, G.S.; Murray, D.; Bremner, W.F.; Third, J.L.H.C.; Johnson, M.C.; Foley, S.; et al. Altered circadian relationship between serum nitric oxide, carbon dioxide, and uric acid in multiple sclerosis. Chronobiol. Int. 2004, 21, 739–758. [Google Scholar] [CrossRef] [PubMed]
- Torreilles, F.; Salman-Tabcheh, S.; Guérin, M.; Torreilles, J. Neurodegenerative disorders: The role of peroxynitrite. Brain Res. Brain Res. Rev. 1999, 30, 153–163. [Google Scholar] [CrossRef]
- Parkinson Study Group SURE-PD3 Investigators; Schwarzschild, M.A.; Ascherio, A.; Casaceli, C.; Curhan, G.C.; Fitzgerald, R.; Kamp, C.; Lungu, C.; Macklin, E.A.; Marek, K.; et al. Effect of Urate-Elevating Inosine on Early Parkinson Disease Progression: The SURE-PD3 Randomized Clinical Trial. JAMA 2021, 326, 926–939. [Google Scholar] [CrossRef]
- Simon, K.C.; Eberly, S.; Gao, X.; Oakes, D.; Tanner, C.M.; Shoulson, I.; Fahn, S.; Schwarzschild, M.A.; Ascherio, A.; Parkinson Study, G. Mendelian randomization of serum urate and parkinson disease progression. Ann. Neurol. 2014, 76, 862–868. [Google Scholar] [CrossRef]
- Miao, J.; Liu, J.; Xiao, L.; Zheng, J.; Liu, C.; Zhu, Z.; Li, K.; Luo, W. The Single Nucleotide Polymorphism rs1014290 of the SLC2A9 Gene Is Associated with Uric Acid Metabolism in Parkinson’s Disease. Park. Dis. 2017, 2017, 7184927. [Google Scholar] [CrossRef]
- Ye, B.S.; Lee, W.W.; Ham, J.H.; Lee, J.J.; Lee, P.H.; Sohn, Y.H.; Alzheimer’s Disease Neuroimaging Initiative. Does serum uric acid act as a modulator of cerebrospinal fluid Alzheimer’s disease biomarker related cognitive decline? Eur. J. Neurol. 2016, 23, 948–957. [Google Scholar] [CrossRef]
- Chen, C.; Li, X.; Lv, Y.; Yin, Z.; Zhao, F.; Liu, Y.; Li, C.; Ji, S.; Zhou, J.; Wei, Y.; et al. High Blood Uric Acid Is Associated With Reduced Risks of Mild Cognitive Impairment Among Older Adults in China: A 9-Year Prospective Cohort Study. Front. Aging Neurosci. 2021, 13, 747686. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, J.; Zeng, J.; He, Y. Relationship between serum uric acid level and mild cognitive impairment in Chinese community elderly. BMC Neurol. 2017, 17, 146. [Google Scholar] [CrossRef] [PubMed]
- Pellecchia, M.T.; Savastano, R.; Moccia, M.; Picillo, M.; Siano, P.; Erro, R.; Vallelunga, A.; Amboni, M.; Vitale, C.; Santangelo, G.; et al. Lower serum uric acid is associated with mild cognitive impairment in early Parkinson’s disease: A 4-year follow-up study. J. Neural Transm. 2016, 123, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Vannorsdall, T.D.; Kueider, A.M.; Carlson, M.C.; Schretlen, D.J. Higher baseline serum uric acid is associated with poorer cognition but not rates of cognitive decline in women. Exp. Gerontol. 2014, 60, 136–139. [Google Scholar] [CrossRef]
- Xue, L.; Liu, Y.; Xue, H.; Xue, J.; Sun, K.; Wu, L.; Hou, P. Low uric acid is a risk factor in mild cognitive impairment. Neuropsychiatr. Dis. Treat. 2017, 13, 2363–2367. [Google Scholar] [CrossRef] [PubMed]
- Latourte, A.; Bardin, T.; Richette, P. Uric acid and cognitive decline: A double-edge sword? Curr. Opin. Rheumatol. 2018, 30, 183–187. [Google Scholar] [CrossRef]
- Gosling, A.L.; Matisoo-Smith, E.; Merriman, T.R. Hyperuricaemia in the Pacific: Why the elevated serum urate levels? Rheumatol. Int. 2014, 34, 743–757. [Google Scholar] [CrossRef]
- Simmonds, H.A.; McBride, M.B.; Hatfield, P.J.; Graham, R.; McCaskey, J.; Jackson, M. Polynesian women are also at risk for hyperuricaemia and gout because of a genetic defect in renal urate handling. Rheumatology 1994, 33, 932–937. [Google Scholar] [CrossRef]
- Healey, L.A.; Jones, K.W. Hyperuricemia in American Samoans. Arthritis Rheum. 1971, 14, 283–285. [Google Scholar] [CrossRef]
- Adams, W.H.; Harper, J.A.; Heotis, P.M.; Jamner, A.H. Hyperuricemia in the inhabitants of the Marshall Islands. Arthritis Rheum. 1984, 27, 713–716. [Google Scholar] [CrossRef]
- Ghaemi-Oskouie, F.; Shi, Y. The role of uric acid as an endogenous danger signal in immunity and inflammation. Curr. Rheumatol. Rep. 2011, 13, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Roman, Y.; Tiirikainen, M.; Prom-Wormley, E. The prevalence of the gout-associated polymorphism rs2231142 G>T in ABCG2 in a pregnant female Filipino cohort. Clin. Rheumatol. 2020, 39, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Oral, A.; Sahin, T.; Turker, F.; Kocak, E. Relationship Between Serum Uric Acid Levels and Nonalcoholic Fatty Liver Disease in Non-Obese Patients. Medicina 2019, 55, 600. [Google Scholar] [CrossRef]
- Kanbay, M.; Jensen, T.; Solak, Y.; Le, M.; Roncal-Jimenez, C.; Rivard, C.; Lanaspa, M.A.; Nakagawa, T.; Johnson, R.J. Uric acid in metabolic syndrome: From an innocent bystander to a central player. Eur. J. Intern. Med. 2016, 29, 3–8. [Google Scholar] [CrossRef]
- Soletsky, B.; Feig, D.I. Uric acid reduction rectifies prehypertension in obese adolescents. Hypertension 2012, 60, 1148–1156. [Google Scholar] [CrossRef]
- Feig, D.I.; Soletsky, B.; Johnson, R.J. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: A randomized trial. JAMA 2008, 300, 924–932. [Google Scholar] [CrossRef]
- Assadi, F. Allopurinol enhances the blood pressure lowering effect of enalapril in children with hyperuricemic essential hypertension. J. Nephrol. 2014, 27, 51–56. [Google Scholar] [CrossRef]
- Kanbay, M.; Ozkara, A.; Selcoki, Y.; Isik, B.; Turgut, F.; Bavbek, N.; Uz, E.; Akcay, A.; Yigitoglu, R.; Covic, A. Effect of treatment of hyperuricemia with allopurinol on blood pressure, creatinine clearence, and proteinuria in patients with normal renal functions. Int. Urol. Nephrol. 2007, 39, 1227–1233. [Google Scholar] [CrossRef]
- Kanbay, M.; Huddam, B.; Azak, A.; Solak, Y.; Kadioglu, G.K.; Kirbas, I.; Duranay, M.; Covic, A.; Johnson, R.J. A randomized study of allopurinol on endothelial function and estimated glomular filtration rate in asymptomatic hyperuricemic subjects with normal renal function. Clin. J. Am. Soc. Nephrol. 2011, 6, 1887–1894. [Google Scholar] [CrossRef]
- Gaffo, A.L.; Calhoun, D.A.; Rahn, E.J.; Oparil, S.; Li, P.; Dudenbostel, T.; Feig, D.I.; Redden, D.T.; Muntner, P.; Foster, P.J.; et al. Effect of Serum Urate Lowering With Allopurinol on Blood Pressure in Young Adults: A Randomized, Controlled, Crossover Trial. Arthritis Rheumatol. 2021, 73, 1514–1522. [Google Scholar] [CrossRef]
- McMullan, C.J.; Borgi, L.; Fisher, N.; Curhan, G.; Forman, J. Effect of Uric Acid Lowering on Renin-Angiotensin-System Activation and Ambulatory BP: A Randomized Controlled Trial. Clin. J. Am. Soc. Nephrol. 2017, 12, 807–816. [Google Scholar] [CrossRef]
- Ortiz-Uriarte, M.; Betancourt-Gaztambide, J.; Perez, A.; Roman, Y.M. Urate-Lowering Therapy Use among US Adults with Gout and the Relationship between Patients’ Gout Treatment Status and Associated Comorbidities. Rheumato 2023, 3, 74–85. [Google Scholar] [CrossRef]
- Parkinson Study Group SURE-PD Investigators; Schwarzschild, M.A.; Ascherio, A.; Beal, M.F.; Cudkowicz, M.E.; Curhan, G.C.; Hare, J.M.; Hooper, D.C.; Kieburtz, K.D.; Macklin, E.A.; et al. Inosine to increase serum and cerebrospinal fluid urate in Parkinson disease: A randomized clinical trial. JAMA Neurol. 2014, 71, 141–150. [Google Scholar] [CrossRef]
- Nicholson, K.; Chan, J.; Macklin, E.A.; Levine-Weinberg, M.; Breen, C.; Bakshi, R.; Grasso, D.L.; Wills, A.-M.; Jahandideh, S.; Taylor, A.A.; et al. Pilot trial of inosine to elevate urate levels in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Walk, D.; Nicholson, K.; Locatelli, E.; Chan, J.; Macklin, E.A.; Ferment, V.; Manousakis, G.; Chase, M.; Connolly, M.; Dagostino, D.; et al. Randomized trial of inosine for urate elevation in amyotrophic lateral sclerosis. Muscle Nerve 2023, 67, 378–386. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Possible Functions | References |
|---|---|---|---|
| ABCG2 | ATP binding cassette subfamily G member 2 (ABCG2) | Regulating renal and gut excretion of urate. Gene polymorphisms have been strongly linked to urate underexcretion and the risk of early-onset gout in men. Genetic polymorphisms may also influence the therapeutic response to allopurinol and rosuvastatin. | [23,26,27,28] |
| GCKR | Glucokinase regulator (GCKR) | Regulatory protein that inhibits glucokinase in the liver and pancreatic islet cells by forming an inactive complex with the enzyme. Gene polymorphisms have been associated with fasting glucose, maturity-onset type-2 diabetes, hyperuricemia, and gout. | [23,26,28] |
| LRRC16A | Capping protein regulator and myosin 1 linker 1 (CARMIL1) | Cytoskeleton-associated protein. Gene polymorphisms have been associated with urate concentrations and gout subtype. | [23,26] |
| PDZK1 | PDZK domain-containing scaffolding protein | Mediates the localization of cell surface proteins and plays a critical role in cholesterol metabolism. Gene polymorphisms have been linked to dyslipidemia, hyperuricemia, and gout. | [23,24,26,28] |
| SLC2A9 | Solute carrier family 2 member 9 (GLUT9) | Regulating renal uric acid reabsorption. Gene polymorphisms have been linked to the risk of gout, especially in women. | [23,24,26,28] |
| SLC16A9 | Solute carrier family 16 member 9 (MCT9) | Regulating monocarboxylic acid transporter. Gene polymorphisms have been linked to uric acid concentrations. | [23,26] |
| SLC17A1 | Solute carrier family 17 member 1 (NPT1) | Sodium phosphate cotransporter. Gene polymorphisms have been linked with hyperuricemia and gout. | [23,26] |
| SLC22A11 | Solute carrier family 22 member 11 (OAT4) | Urate reabsorption transporter. A target for some uricosuric drugs. Gene polymorphisms have been associated with hyperuricemia. | [23,26] |
| SLC22A12 | Solute carrier family 22 member 12 (URAT1) | Uric acid reabsorption transporter. A major target for uricosuric drugs. Gene polymorphisms have been associated with hyperuricemia and gout. Loss of function in the gene can also lead to hypouricemia. | [23,26] |
| NRXN2 | Neurexin 2 | Member of the neurexin gene family that serves as a cell adhesion molecule. Genetic polymorphisms were associated with urate concentrations in Europeans and Chinese. | [24,26] |
| INHBC | INHBC | Member of the transforming growth factor ß superfamily that may inhibit activin A signaling, affecting a variety of biological functions including pituitary hormone secretion and insulin secretion. | [24,26] |
| HNF1A | Hepatic nuclear factor 1A | Encodes a transcription factor and enhances the promoter activity of PDZK1, URAT1, NPT4, and OAT4 in the human proximal tubule. | [28] |
| HNF4A | Hepatic nuclear factor 4A | Encodes another nuclear receptor and transcription factor that controls the expression of multiple other genes. It was shown to regulate the expression of SLC2A9 and other members of the urate transportome. | [28] |
| HNF4G | Hepatic nuclear factor 4G | Encodes other transcription factors. Genetic polymorphisms are significantly associated with increased urate concentration in Europeans and gout in Chinese. | [24,29] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roman, Y.M. The Role of Uric Acid in Human Health: Insights from the Uricase Gene. J. Pers. Med. 2023, 13, 1409. https://doi.org/10.3390/jpm13091409
Roman YM. The Role of Uric Acid in Human Health: Insights from the Uricase Gene. Journal of Personalized Medicine. 2023; 13(9):1409. https://doi.org/10.3390/jpm13091409
Chicago/Turabian StyleRoman, Youssef M. 2023. "The Role of Uric Acid in Human Health: Insights from the Uricase Gene" Journal of Personalized Medicine 13, no. 9: 1409. https://doi.org/10.3390/jpm13091409
APA StyleRoman, Y. M. (2023). The Role of Uric Acid in Human Health: Insights from the Uricase Gene. Journal of Personalized Medicine, 13(9), 1409. https://doi.org/10.3390/jpm13091409