


Lymantria Dispar Iflavirus 1 RNA Comprises a Large Proportion of RNA in Adult L. dispar Moths




Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA-Seq Data, LdIV1 Contig Assembly, and Genome Sequence Phylogeny
2.2. Polyprotein Sequence Analysis, Codon Alignment Phylogeny, and IRES Prediction
2.3. Assessing LdIV1 Transcript Levels
3. Results
3.1. Genome Sequences of, and Relationships among, LdIV1 Variants from Different Host Spongy Moth Populations
3.2. Polyprotein Sequence Variation and Phylogeny
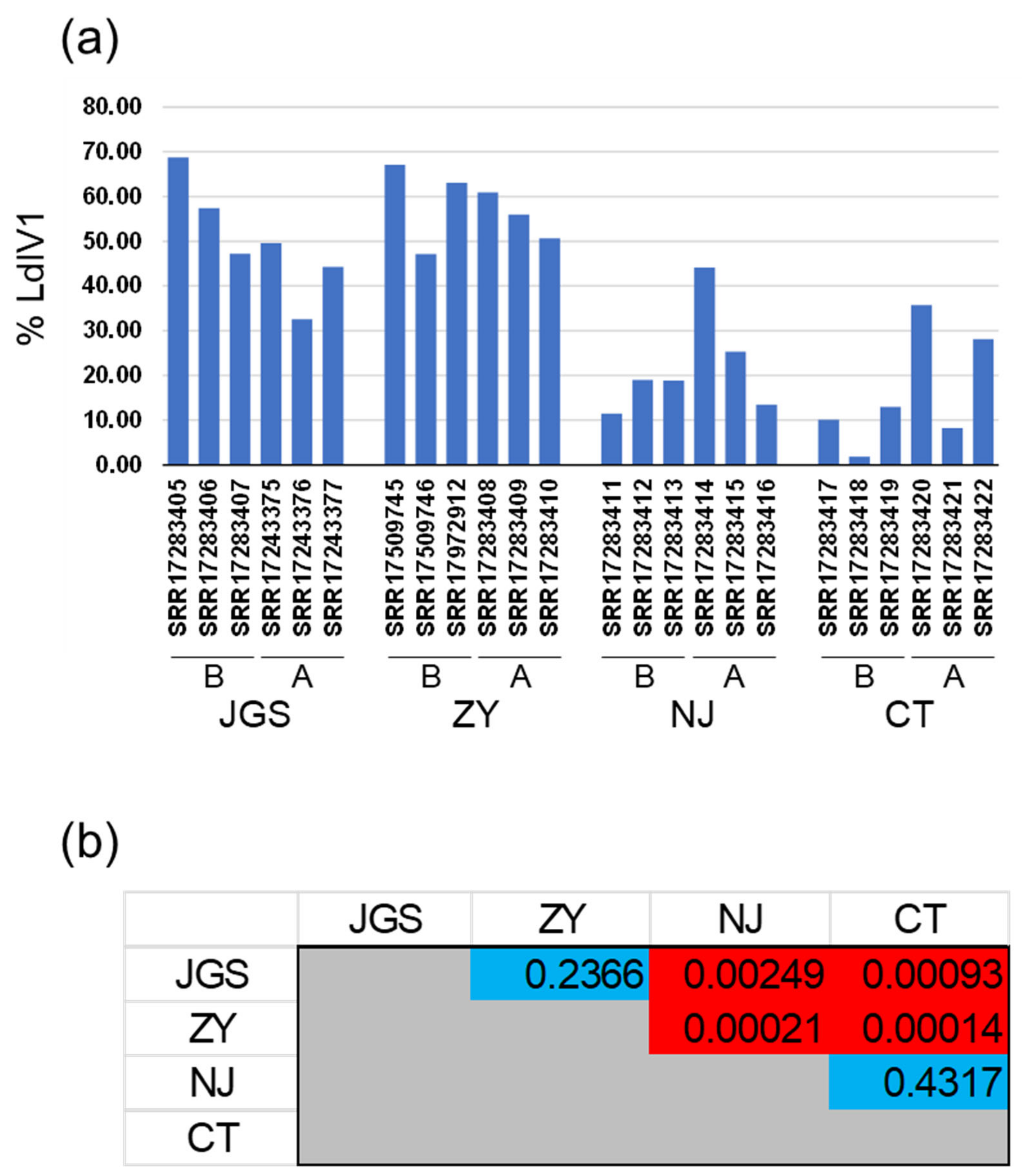
3.3. Steady-State LdIV1 Transcript Levels in Adult Spongy Moths
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, C.-X.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Kang, Y.-J.; Chen, L.-J.; Qin, X.-C.; Xu, J.; Holmes, E.C.; Zhang, Y.-Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhang, Y.-Z.; Holmes, E.C. Meta-transcriptomics and the evolutionary biology of RNA viruses. Virus Res. 2018, 243, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Valles, S.M.; Chen, Y.; Firth, A.; Guérin, D.M.A.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Iflaviridae. J. Gen. Virol. 2017, 98, 527–528. [Google Scholar] [CrossRef]
- De Miranda, J.R.; Genersch, E. Deformed wing virus. J. Invertebr. Pathol. 2010, 103 (Suppl. S1), S48–S61. [Google Scholar] [CrossRef]
- Virto, C.; Navarro, D.; Tellez, M.M.; Herrero, S.; Williams, T.; Murillo, R.; Caballero, P. Natural populations of Spodoptera exigua are infected by multiple viruses that are transmitted to their offspring. J. Invertebr. Pathol. 2014, 122, 22–27. [Google Scholar] [CrossRef]
- Bailey, L.; Gibbs, A.; Woods, R. Sacbrood virus of the larval honey bee (Apis mellifera linnaeus). Virology 1964, 23, 425–429. [Google Scholar] [CrossRef]
- Li, J.; Wang, T.; Evans, J.D.; Rose, R.; Zhao, Y.; Li, Z.; Li, J.; Huang, S.; Heerman, M.; Rodríguez-García, C.; et al. The Phylogeny and Pathogenesis of Sacbrood Virus (SBV) Infection in European Honey Bees, Apis mellifera. Viruses 2019, 11, 61. [Google Scholar] [CrossRef]
- Geng, P.; Li, W.; Lin, L.; de Miranda, J.R.; Emrich, S.; An, L.; Terenius, O. Genetic Characterization of a Novel Iflavirus Associated with Vomiting Disease in the Chinese Oak Silkmoth Antheraea pernyi. PLoS ONE 2014, 9, e92107. [Google Scholar] [CrossRef]
- Perera, O.P.; Snodgrass, G.L.; Allen, K.C.; Jackson, R.E.; Becnel, J.J.; O’leary, P.F.; Luttrell, R.G. The complete genome sequence of a single-stranded RNA virus from the tarnished plant bug, Lygus lineolaris (Palisot de Beauvois). J. Invertebr. Pathol. 2012, 109, 11–19. [Google Scholar] [CrossRef]
- Sparks, M.E.; Gundersen-Rindal, D.E.; Harrison, R.L. Complete Genome Sequence of a Novel Iflavirus from the Transcriptome of Halyomorpha halys, the Brown Marmorated Stink Bug. Genome Announc. 2013, 1, e00910-13. [Google Scholar] [CrossRef]
- Carrillo-Tripp, J.; Krueger, E.N.; Harrison, R.; Toth, A.L.; Miller, W.A.; Bonning, B.C. Lymantria dispar iflavirus 1 (LdIV1), a new model to study iflaviral persistence in lepidopterans. J. Gen. Virol. 2014, 95, 2285–2296. [Google Scholar] [CrossRef]
- Pogue, M.G.; Schaefer, P.W. A Review of Selected Species of Lymantria Hubner [1819] (Lepidoptera: Noctuidae: Lymantriinae) from Subtropical and Temperate Regions of Asia, including the Descriptions of Three New Species, Some Potentially Invasive to North America; Forest Health Technology Enterprise Team: Juneau, AK, USA, 2007. [Google Scholar]
- Wang, Y.-M.; Sparks, M.E.; Harrison, R.L.; Shi, J. Analyses of adult transcriptomes from four different populations of the spongy moth, Lymantria dispar L., from China and the USA. Sci. Rep. 2022, 12, 18232. [Google Scholar] [CrossRef]
- Pavlushin, S.V.; Ilinsky, Y.Y.; Belousova, I.A.; Bayborodin, S.I.; Lunev, E.A.; Kechin, A.A.; Khrapov, E.A.; Filipenko, M.L.; Toshchakov, S.V.; Martemyanov, V.V. Appearances are deceptive: Three RNA viruses co-infected with the nucleopolyhedrovirus in host Lymantria dispar. Virus Res. 2021, 297, 198371. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; Hendrickson, R.C.; et al. Recent changes to virus taxonomy ratified by the International Committee on Taxonomy of Viruses (2022). Arch. Virol. 2022, 167, 2429–2440. [Google Scholar] [CrossRef]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; McVeigh, R.; O’neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef]
- Goldman, N.; Yang, Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994, 11, 725–736. [Google Scholar] [CrossRef]
- Kolekar, P.; Pataskar, A.; Kulkarni-Kale, U.; Pal, J.; Kulkarni, A. IRESPred: Web Server for Prediction of Cellular and Viral Internal Ribosome Entry Site (IRES). Sci. Rep. 2016, 6, 27436. [Google Scholar] [CrossRef]
- Wang, J.; Gribskov, M. IRESpy: An XGBoost model for prediction of internal ribosome entry sites. BMC Bioinform. 2019, 20, 409. [Google Scholar] [CrossRef]
- Ongus, J.R.; Roode, E.C.; Pleij, C.W.A.; Vlak, J.M.; van Oers, M.M. The 5′ non-translated region of Varroa destructor virus 1 (genus Iflavirus): Structure prediction and IRES activity in Lymantria dispar cells. J. Gen. Virol. 2006, 87, 3397–3407. [Google Scholar] [CrossRef]
- Lu, J.; Hu, Y.; Hu, L.; Zong, S.; Cai, D.; Wang, J.; Yu, H.; Zhang, J. Ectropis obliqua picorna-like virus IRES-driven internal initiation of translation in cell systems derived from different origins. J. Gen. Virol. 2007, 88, 2834–2838. [Google Scholar] [CrossRef]
- Harrison, R.L.; Rowley, D.L.; Keena, M.A. Pathology and genome sequence of a Lymantria dispar multiple nucleopolyhedrovirus (LdMNPV) isolate from Heilongjiang, China. J. Invertebr. Pathol. 2020, 177, 107495. [Google Scholar] [CrossRef]
- Picq, S.; Keena, M.; Havill, N.; Stewart, D.; Pouliot, E.; Boyle, B.; Levesque, R.C.; Hamelin, R.C.; Cusson, M. Assessing the potential of genotyping-by-sequencing-derived single nucleotide polymorphisms to identify the geographic origins of intercepted gypsy moth (Lymantria dispar) specimens: A proof-of-concept study. Evol. Appl. 2018, 11, 325–339. [Google Scholar] [CrossRef]
- Goodwin, R.H.; Tompkins, G.J.; McCawley, P. Gypsy moth cell lines divergent in viral susceptibility: I. Culture and identification. In Vitro 1978, 14, 485–494. [Google Scholar] [CrossRef]
- Slavicek, J.M. Temporal analysis and spatial mapping of Lymantria dispar nuclear polyhedrosis virus transcripts and in vitro translation polypeptides. Virus Res. 1991, 20, 223–236. [Google Scholar] [CrossRef]
- Kuzioa, J.; Pearson, M.N.; Harwood, S.H.; Funk, C.; Evans, J.; Slavicek, J.M.; Rohrmann, G.F. Sequence and Analysis of the Genome of a Baculovirus Pathogenic for Lymantria dispar. Virology 1999, 253, 17–34. [Google Scholar] [CrossRef]
- Harrison, R.L.; Keena, M.A.; Rowley, D.L. Classification, genetic variation and pathogenicity of Lymantria dispar nucleopolyhedrovirus isolates from Asia, Europe, and North America. J. Invertebr. Pathol. 2014, 116, 27–35. [Google Scholar] [CrossRef]
- Martemyanov, V.; Podgwaite, J.; Belousova, I.; Pavlushin, S.; Slavicek, J.; Baturina, O.; Kabilov, M.; Ilyinykh, A. A comparison of the adaptations of strains of Lymantria dispar multiple nucleopolyhedrovirus to hosts from spatially isolated populations. J. Invertebr. Pathol. 2017, 146, 41–46. [Google Scholar] [CrossRef]
- Lozano, G.; Fernandez, N.; Martinez-Salas, E. Modeling Three-Dimensional Structural Motifs of Viral IRES. J. Mol. Biol. 2016, 428, 767–776. [Google Scholar] [CrossRef]
- Zhang, Y.-Z.; Chen, Y.-M.; Wang, W.; Qin, X.-C.; Holmes, E.C. Expanding the RNA Virosphere by Unbiased Metagenomics. Annu. Rev. Virol. 2019, 6, 119–139. [Google Scholar] [CrossRef]
- Jakubowska, A.K.; Murillo, R.; Carballo, A.; Williams, T.; van Lent, J.W.; Caballero, P.; Herrero, S. Iflavirus increases its infectivity and physical stability in association with baculovirus. PeerJ 2016, 4, e1687. [Google Scholar] [CrossRef] [PubMed]
- Carballo, A.; Murillo, R.; Jakubowska, A.; Herrero, S.; Williams, T.; Caballero, P. Co-infection with iflaviruses influences the insecticidal properties of Spodoptera exigua multiple nucleopolyhedrovirus occlusion bodies: Implications for the production and biosecurity of baculovirus insecticides. PLoS ONE 2017, 12, e0177301. [Google Scholar] [CrossRef] [PubMed]
- Carballo, A.; Williams, T.; Murillo, R.; Caballero, P. Iflavirus Covert Infection Increases Susceptibility to Nucleopolyhedrovirus Disease in Spodoptera exigua. Viruses 2020, 12, 509. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.C.; Ahmed, S.; Mollah, M.I.; Kim, M.I.M.A.Y. Antiviral Treatment Reveals a Cooperative Pathogenicity of Baculovirus and Iflavirus in Spodoptera exigua, a Lepidopteran Insect. J. Microbiol. Biotechnol. 2021, 31, 529–539. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Spongy Moth Population | Time of RNA Harvest | SRA Identifiers |
|---|---|---|
| Jingeshan, Hebei, China (JGS) | Before mating | SRR17283405, SRR17283406, SRR17283407 |
| After mating and oviposition | SRR17243375, SRR17243376, SRR17243377 | |
| Zunyi, Guizhou, China (ZY) | Before mating | SRR17509745, SRR17509746, SRR17972912 |
| After mating and oviposition | SRR17283408, SRR17283409, SRR17283410 | |
| New Jersey, USA (NJ) | Before mating | SRR17283411, SRR17283412, SRR17283413 |
| After mating and oviposition | SRR17283414, SRR17283415, SRR17283416 | |
| Connecticut, USA (CT) | Before mating | SRR17283417, SRR17283418, SRR17283419 |
| After mating and oviposition | SRR17283420, SRR17283421, SRR17283422 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sparks, M.E.; Wang, Y.-M.; Shi, J.; Harrison, R.L. Lymantria Dispar Iflavirus 1 RNA Comprises a Large Proportion of RNA in Adult L. dispar Moths. Insects 2023, 14, 466. https://doi.org/10.3390/insects14050466
Sparks ME, Wang Y-M, Shi J, Harrison RL. Lymantria Dispar Iflavirus 1 RNA Comprises a Large Proportion of RNA in Adult L. dispar Moths. Insects. 2023; 14(5):466. https://doi.org/10.3390/insects14050466
Chicago/Turabian StyleSparks, Michael E., Yi-Ming Wang, Juan Shi, and Robert L. Harrison. 2023. "Lymantria Dispar Iflavirus 1 RNA Comprises a Large Proportion of RNA in Adult L. dispar Moths" Insects 14, no. 5: 466. https://doi.org/10.3390/insects14050466
APA StyleSparks, M. E., Wang, Y. -M., Shi, J., & Harrison, R. L. (2023). Lymantria Dispar Iflavirus 1 RNA Comprises a Large Proportion of RNA in Adult L. dispar Moths. Insects, 14(5), 466. https://doi.org/10.3390/insects14050466