
Switch-Like Roles for Polycomb Proteins from Neurodevelopment to Neurodegeneration

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. A Snapshot of Polycomb Group Proteins
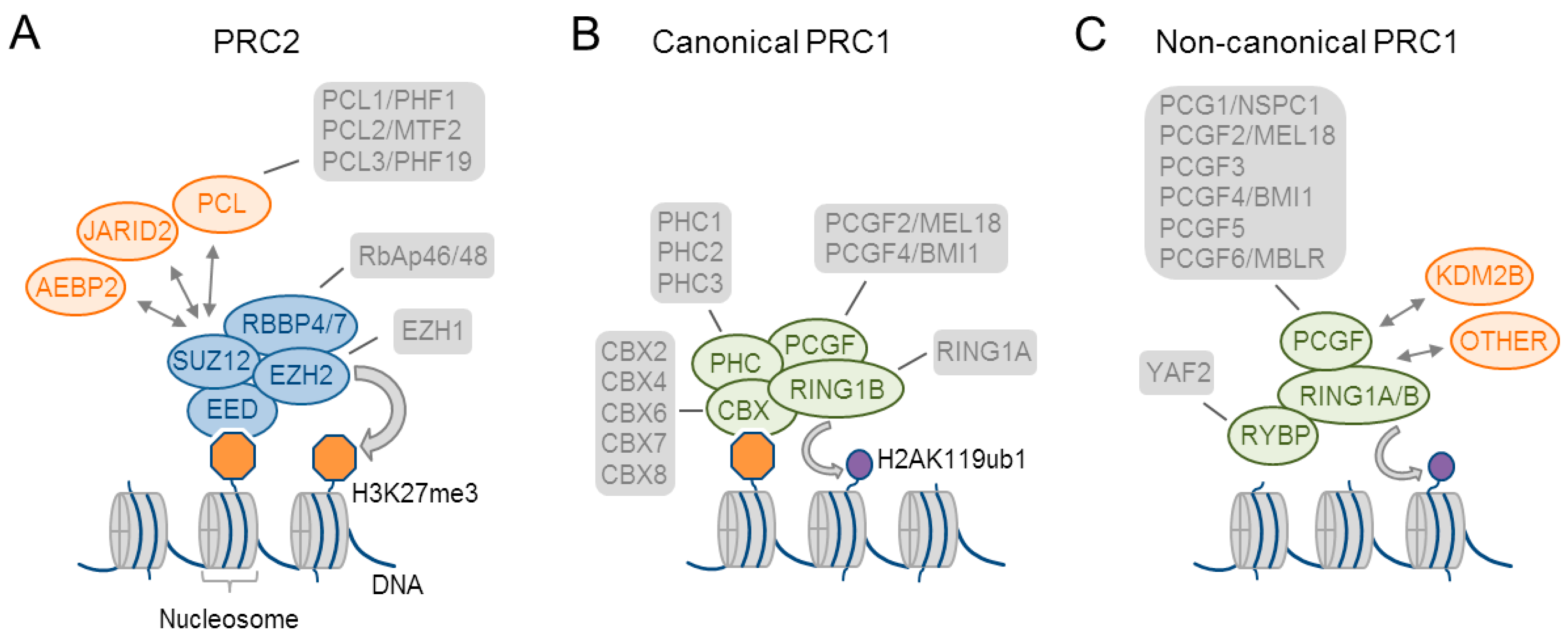
2.1. Composition of Polycomb Group Complexes
2.2. Recruitment of Polycomb Group Complexes
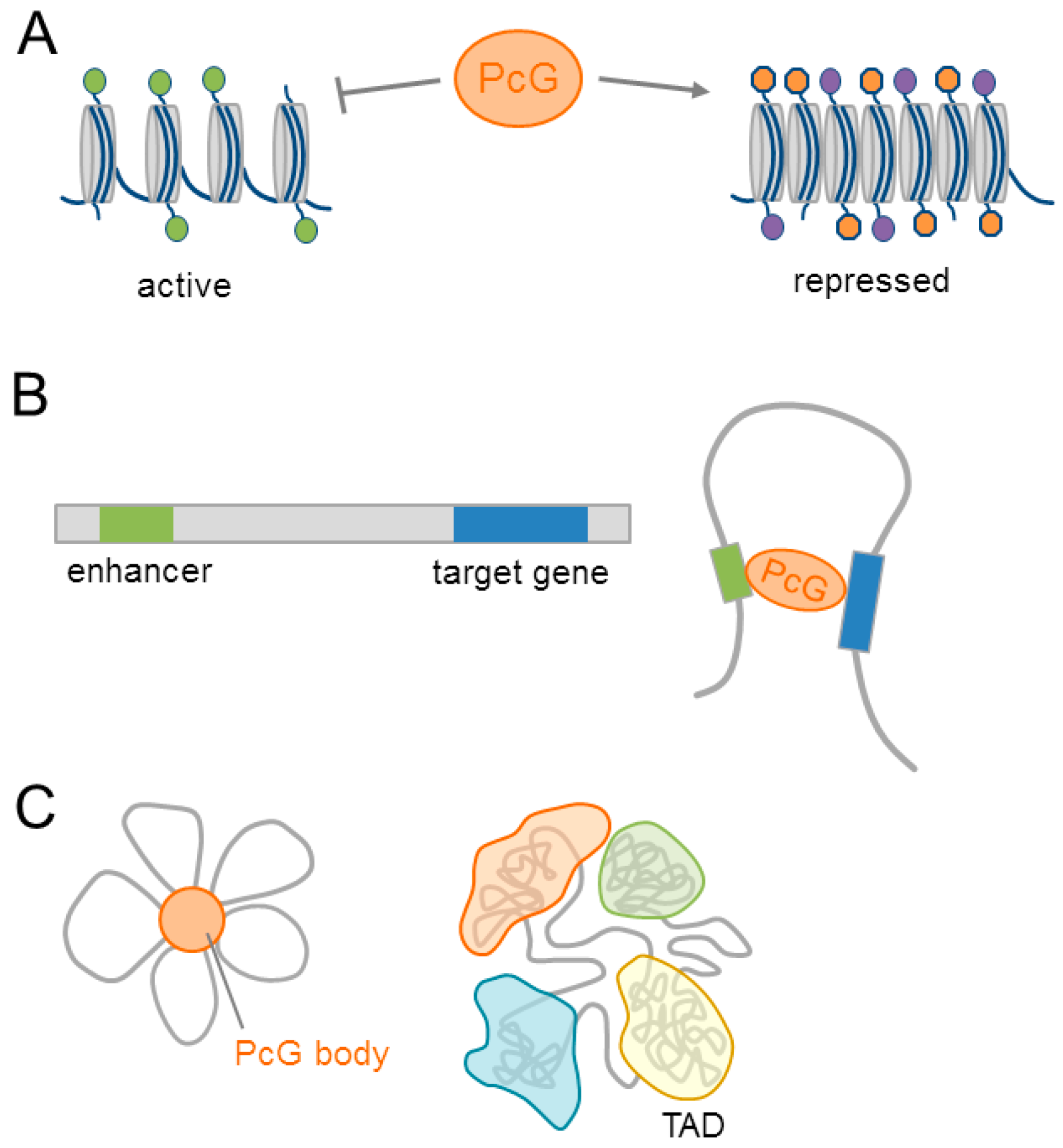
2.3. Polycomb Group Complexes Regulate Chromatin at Different Scales
3. Embryonic and Adult Neurogenesis
4. Polycomb Group Complexes Guide Dynamic Transitions in Neurodevelopment
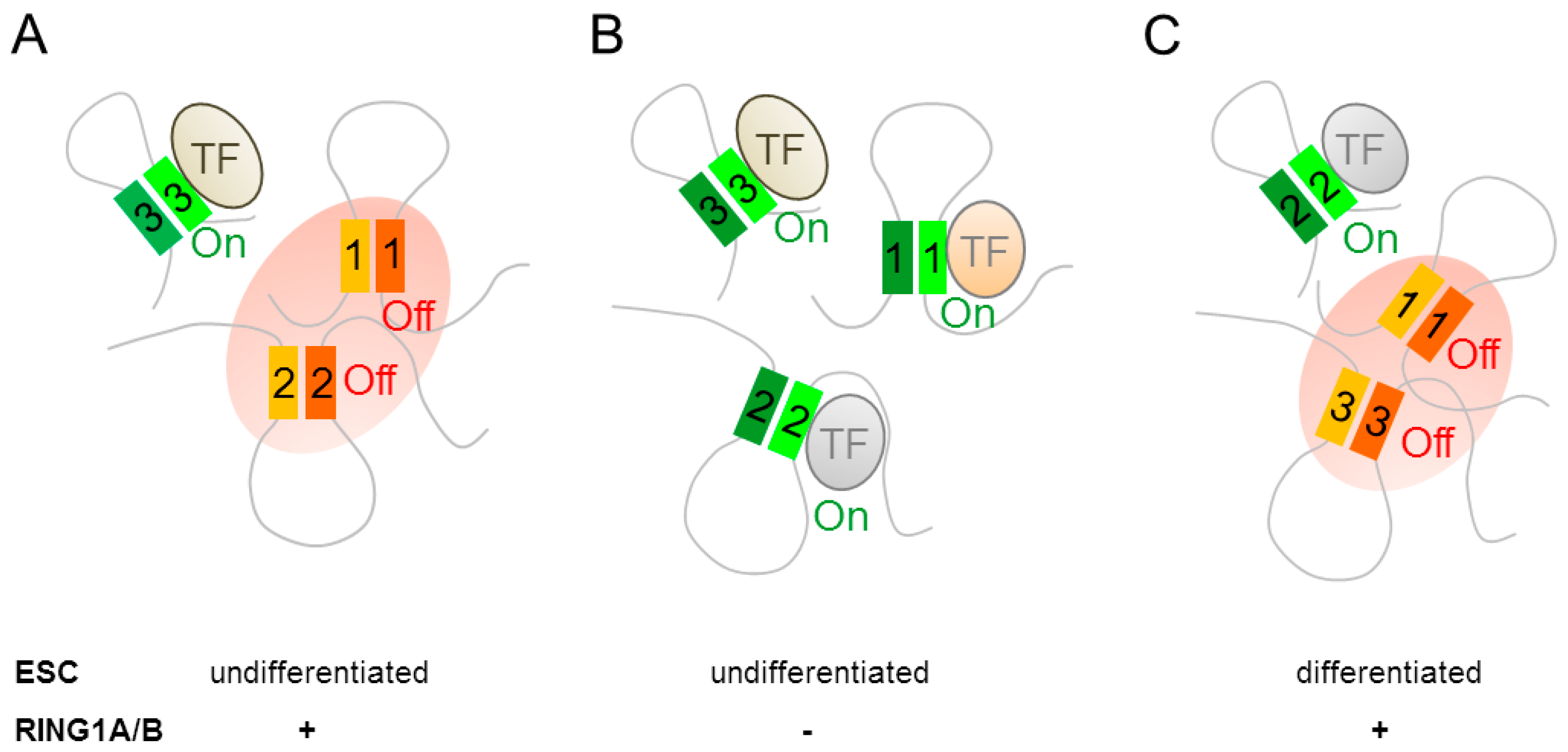
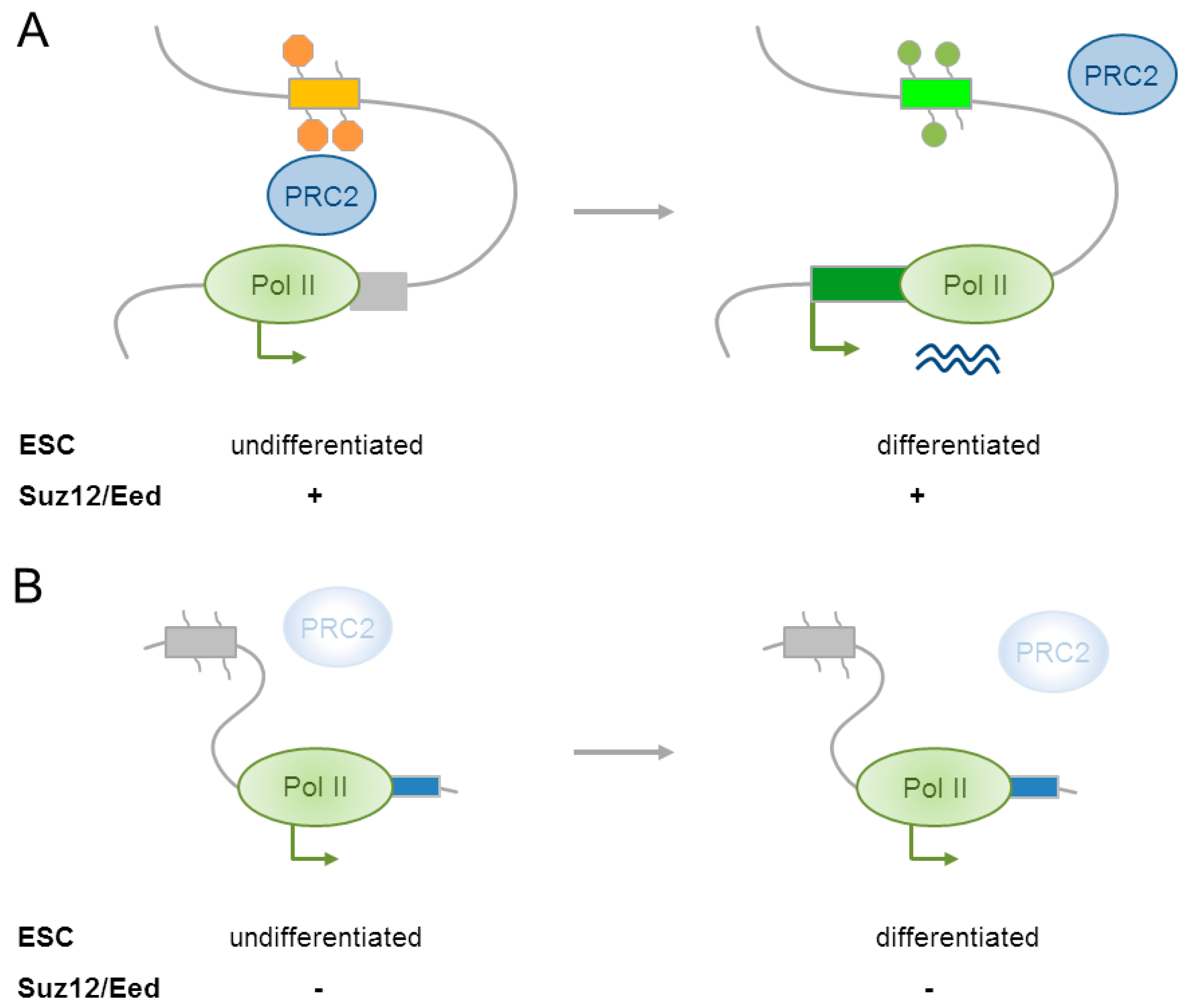
4.1. Polycomb Group Complexes in Embryonic Stem Cells Restrain Neural Differentiation
4.2. Polycomb Group Complexes in Neural Progenitor Cells
4.3. Polycomb Group Complexes in Differentiating Neural Cell Types
4.4. Polycomb Group Complexes in Adult NSCs
4.5. Polycomb Group Complexes in Fate Restricted Neuronal Precursors and Differentiating Neurons
4.6. Polycomb Group Complexes in Mature Neurons and Neurodegenerative Disease
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Duncan, I.M. Polycomblike: A gene that appears to be required for the normal expression of the bithorax and antennapedia gene complexes of Drosophila melanogaster. Genetics 1982, 102, 49–70. [Google Scholar] [PubMed]
- Lewis, E.B. A gene complex controlling segmentation in Drosophila. Nature 1978, 276, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Kingston, R.E. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Ringrose, L.; Paro, R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu. Rev. Genet. 2004, 38, 413–443. [Google Scholar] [CrossRef] [PubMed]
- Capecchi, M.R. Hox genes and mammalian development. Cold Spring Harb. Symp. Quant. Biol. 1997, 62, 273–281. [Google Scholar] [PubMed]
- Nüsslein-Volhard, C. Of flies and fishes. Science 1994, 266, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Corley, M.; Kroll, K.L. The roles and regulation of Polycomb complexes in neural development. Cell Tissue Res. 2015, 359, 65–85. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, A.; Magnuson, T. Murine Polycomb- and trithorax-group genes regulate homeotic pathways and beyond. Trends Genet. 1997, 13, 167–170. [Google Scholar] [CrossRef]
- Zakany, J.; Duboule, D. The role of Hox genes during vertebrate limb development. Curr. Opin. Genet. Dev. 2007, 17, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Tanay, A.; O’Donnell, A.H.; Damelin, M.; Bestor, T.H. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc. Natl. Acad. Sci. USA 2007, 104, 5521–5526. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Molina, S.; Respuela, P.; Tebartz, C.; Kolovos, P.; Nikolic, M.; Fueyo, R.; van Ijcken, W.F.J.; Grosveld, F.; Frommolt, P.; Bazzi, H.; et al. PRC2 Facilitates the Regulatory Topology Required for Poised Enhancer Function during Pluripotent Stem Cell Differentiation. Cell Stem Cell 2017, 20, 689–705.e9. [Google Scholar] [CrossRef] [PubMed]
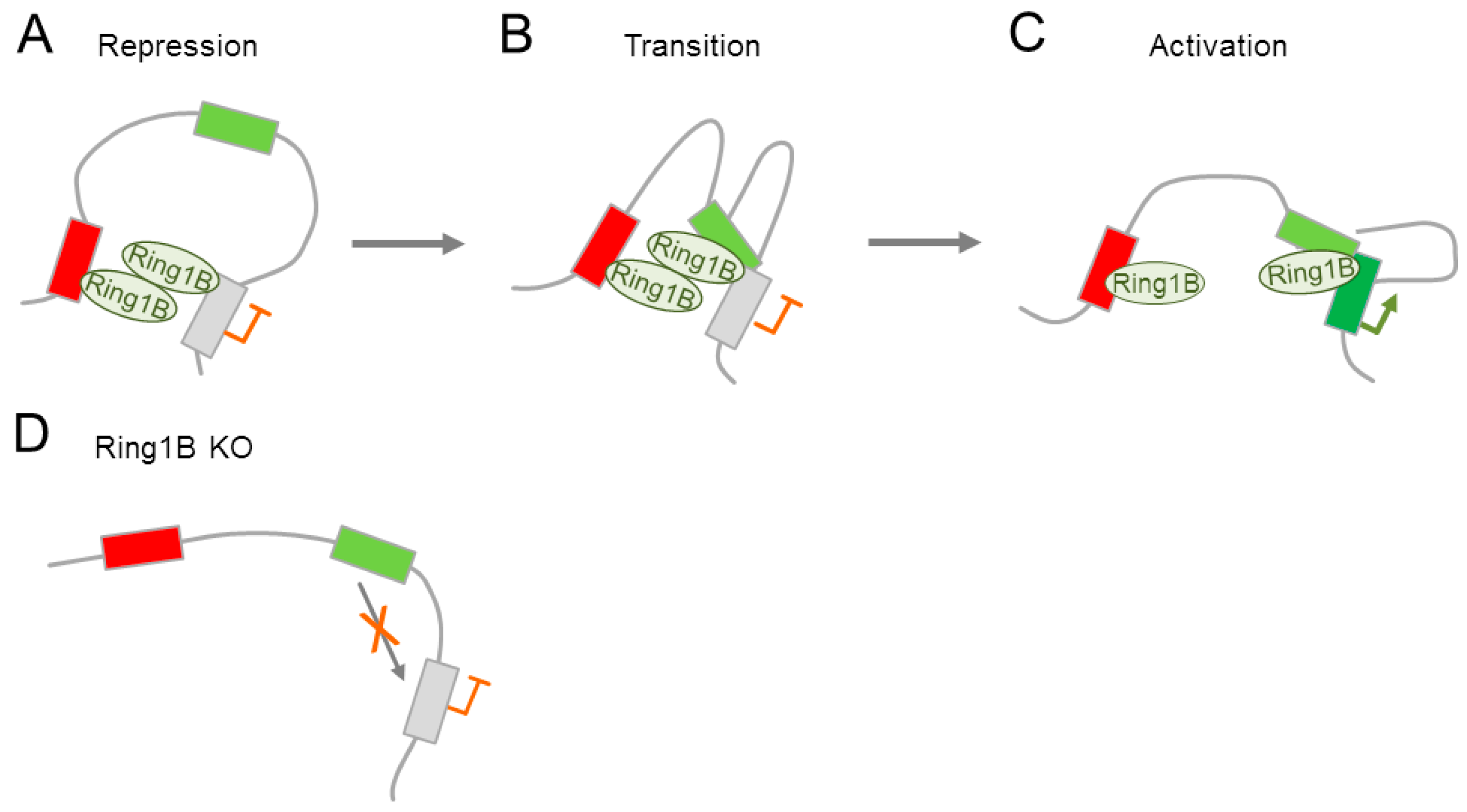
- Kondo, T.; Isono, K.; Kondo, K.; Endo, T.A.; Itohara, S.; Vidal, M.; Koseki, H. Polycomb potentiates Meis2 activation in midbrain by mediating interaction of the promoter with a tissue-specific enhancer. Dev. Cell 2014, 28, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schübeler, D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 2008, 30, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Zimmermann, C.A.; Spengler, D. Molecular epigenetic switches in neurodevelopment in health and disease. Front. Behav. Neurosci. 2015, 9, 120. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Bourbon, H.-M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shao, Z.; Li, D.; Xie, H.; Kim, W.; Huang, J.; Taylor, J.E.; Pinello, L.; Glass, K.; Jaffe, J.D.; et al. Developmental control of polycomb subunit composition by GATA factors mediates a switch to non-canonical functions. Mol. Cell 2015, 57, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, K.; Zare, H.; Wang, A.H.; Sartorelli, V. Polycomb protein Ezh1 promotes RNA polymerase II elongation. Mol. Cell 2012, 45, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Hauri, S.; Comoglio, F.; Seimiya, M.; Gerstung, M.; Glatter, T.; Hansen, K.; Aebersold, R.; Paro, R.; Gstaiger, M.; Beisel, C. A High-Density Map for Navigating the Human Polycomb Complexome. Cell Rep. 2016, 17, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Trupke, J.; Ringrose, L. The quest for mammalian Polycomb response elements: Are we there yet? Chromosoma 2016, 125, 471–496. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Ku, M.; Koche, R.P.; Rheinbay, E.; Mendenhall, E.M.; Endoh, M.; Mikkelsen, T.S.; Presser, A.; Nusbaum, C.; Xie, X.; Chi, A.S.; et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008, 4, e1000242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, N.; Lerdrup, M.; Landt, E.; Agrawal-Singh, S.; Bak, M.; Tommerup, N.; Rappsilber, J.; Södersten, E.; Hansen, K. REST-mediated recruitment of polycomb repressor complexes in mammalian cells. PLoS Genet. 2012, 8, e1002494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hekimoglu-Balkan, B.; Aszodi, A.; Heinen, R.; Jaritz, M.; Ringrose, L. Intergenic Polycomb target sites are dynamically marked by non-coding transcription during lineage commitment. RNA Biol. 2012, 9, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Farcas, A.M.; Blackledge, N.P.; Sudbery, I.; Long, H.K.; McGouran, J.F.; Rose, N.R.; Lee, S.; Sims, D.; Cerase, A.; Sheahan, T.W.; et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. eLife 2012, 1, e00205. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Johansen, J.V.; Helin, K. Fbxl10/Kdm2b recruits polycomb repressive complex 1 to CpG islands and regulates H2A ubiquitylation. Mol. Cell 2013, 49, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Shen, L.; Wan, M.; Taranova, O.; Wu, H.; Zhang, Y. Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat. Cell Biol. 2013, 15, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Cooper, S.; Farcas, A.M.; Blackledge, N.P.; Brockdorff, N. Chromatin sampling—An emerging perspective on targeting polycomb repressor proteins. PLoS Genet. 2013, 9, e1003717. [Google Scholar] [CrossRef] [PubMed]
- Riising, E.M.; Comet, I.; Leblanc, B.; Wu, X.; Johansen, J.V.; Helin, K. Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol. Cell 2014, 55, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Margueron, R.; Ku, M.; Chambon, P.; Bernstein, B.E.; Reinberg, D. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 2010, 24, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Landeira, D.; Sauer, S.; Poot, R.; Dvorkina, M.; Mazzarella, L.; Jørgensen, H.F.; Pereira, C.F.; Leleu, M.; Piccolo, F.M.; Spivakov, M.; et al. Jarid2 is a PRC2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of PRC1 and RNA Polymerase II to developmental regulators. Nat. Cell Biol. 2010, 12, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Cloos, P.A.C.; Walfridsson, J.; Olsson, L.; Bukowski, J.-P.; Johansen, J.V.; Bak, M.; Tommerup, N.; Rappsilber, J.; Helin, K. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature 2010, 464, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.C.; Valouev, A.; Swigut, T.; Zhang, J.; Zhao, Y.; Sidow, A.; Wysocka, J. Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell 2009, 139, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.T.C.; Müller, C.W.; Vermeulen, M.; Müller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.H.; Bracken, A.P.; Pasini, D.; Dietrich, N.; Gehani, S.S.; Monrad, A.; Rappsilber, J.; Lerdrup, M.; Helin, K. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 2008, 10, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Justin, N.; Ohno, K.; Sharpe, M.L.; Son, J.; Drury, W.J.; Voigt, P.; Martin, S.R.; Taylor, W.R.; De Marco, V.; et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 2009, 461, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Rojas, C.; Hernandez, A.J.; Sarma, K.; Lee, J.T. Regulatory interactions between RNA and polycomb repressive complex. Mol. Cell 2014, 55, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Son, J.; Bonasio, R.; Shen, S.S.; Reinberg, D. Nascent RNA interaction keeps PRC2 activity poised and in check. Genes Dev. 2014, 28, 1983–1988. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N. Noncoding RNA and Polycomb recruitment. RNA 2013, 19, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Chopra, V.S.; Hendrix, D.A.; Core, L.J.; Tsui, C.; Lis, J.T.; Levine, M. The polycomb group mutant esc leads to augmented levels of paused Pol II in the Drosophila embryo. Mol. Cell 2011, 42, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Tie, F.; Banerjee, R.; Fu, C.; Stratton, C.A.; Fang, M.; Harte, P.J. Polycomb inhibits histone acetylation by CBP by binding directly to its catalytic domain. Proc. Natl. Acad. Sci. USA 2016, 113, E744–E753. [Google Scholar] [CrossRef] [PubMed]
- Lanzuolo, C.; Roure, V.; Dekker, J.; Bantignies, F.; Orlando, V. Polycomb response elements mediate the formation of chromosome higher-order structures in the bithorax complex. Nat. Cell Biol. 2007, 9, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Cheutin, T.; Cavalli, G. Polycomb silencing: from linear chromatin domains to 3D chromosome folding. Curr. Opin. Genet. Dev. 2014, 25, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Vietri Rudan, M.; Barrington, C.; Henderson, S.; Ernst, C.; Odom, D.T.; Tanay, A.; Hadjur, S. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 2015, 10, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Dowen, J.M.; Fan, Z.P.; Hnisz, D.; Ren, G.; Abraham, B.J.; Zhang, L.N.; Weintraub, A.S.; Schuijers, J.; Lee, T.I.; Zhao, K.; et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 2014, 159, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Isono, K.; Endo, T.A.; Ku, M.; Yamada, D.; Suzuki, R.; Sharif, J.; Ishikura, T.; Toyoda, T.; Bernstein, B.E.; Koseki, H. SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev. Cell 2013, 26, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Gambetta, M.C.; Müller, J. O-GlcNAcylation prevents aggregation of the Polycomb group repressor polyhomeotic. Dev. Cell 2014, 31, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.; Zhuang, X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 2016, 529, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Denholtz, M.; Bonora, G.; Chronis, C.; Splinter, E.; de Laat, W.; Ernst, J.; Pellegrini, M.; Plath, K. Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell 2013, 13, 602–616. [Google Scholar] [CrossRef] [PubMed]
- Joshi, O.; Wang, S.-Y.; Kuznetsova, T.; Atlasi, Y.; Peng, T.; Fabre, P.J.; Habibi, E.; Shaik, J.; Saeed, S.; Handoko, L.; et al. Dynamic Reorganization of Extremely Long-Range Promoter-Promoter Interactions between Two States of Pluripotency. Cell Stem Cell 2015, 17, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.-L.; Song, H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Kriegstein, A.; Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef] [PubMed]
- Götz, M.; Huttner, W.B. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Gage, F.H.; Temple, S. Neural stem cells: generating and regenerating the brain. Neuron 2013, 80, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Meshorer, E.; Yellajoshula, D.; George, E.; Scambler, P.J.; Brown, D.T.; Misteli, T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell 2006, 10, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Young, R.A. Control of the embryonic stem cell state. Cell 2011, 144, 940–954. [Google Scholar] [CrossRef] [PubMed]
- Azuara, V.; Perry, P.; Sauer, S.; Spivakov, M.; Jørgensen, H.F.; John, R.M.; Gouti, M.; Casanova, M.; Warnes, G.; Merkenschlager, M.; et al. Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 2006, 8, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.-K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Alder, O.; Lavial, F.; Helness, A.; Brookes, E.; Pinho, S.; Chandrashekran, A.; Arnaud, P.; Pombo, A.; O’Neill, L.; Azuara, V. Ring1B and Suv39h1 delineate distinct chromatin states at bivalent genes during early mouse lineage commitment. Development 2010, 137, 2483–2492. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Zang, C.; Roh, T.-Y.; Schones, D.E.; Childs, R.W.; Peng, W.; Zhao, K. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 2009, 4, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Dahl, J.A.; Reiner, A.H.; Klungland, A.; Wakayama, T.; Collas, P. Histone H3 lysine 27 methylation asymmetry on developmentally-regulated promoters distinguish the first two lineages in mouse preimplantation embryos. PLoS ONE 2010, 5, e9150. [Google Scholar] [CrossRef] [PubMed]
- Rugg-Gunn, P.J.; Cox, B.J.; Ralston, A.; Rossant, J. Distinct histone modifications in stem cell lines and tissue lineages from the early mouse embryo. Proc. Natl. Acad. Sci. USA 2010, 107, 10783–10790. [Google Scholar] [CrossRef] [PubMed]
- Vastenhouw, N.L.; Zhang, Y.; Woods, I.G.; Imam, F.; Regev, A.; Liu, X.S.; Rinn, J.; Schier, A.F. Chromatin signature of embryonic pluripotency is established during genome activation. Nature 2010, 464, 922–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, G.; Tian, S.; Nie, J.; Yang, C.; Ruotti, V.; Wei, H.; Jonsdottir, G.A.; Stewart, R.; Thomson, J.A. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 2007, 1, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.D.; Han, X.; Chew, J.L.; Liu, J.; Chiu, K.P.; Choo, A.; Orlov, Y.L.; Sung, W.-K.; Shahab, A.; Kuznetsov, V.A.; et al. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell 2007, 1, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Zeltner, N.; Studer, L. Pluripotent stem cell-based disease modeling: current hurdles and future promise. Curr. Opin. Cell Biol. 2015, 37, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Guenther, M.G.; Frampton, G.M.; Soldner, F.; Hockemeyer, D.; Mitalipova, M.; Jaenisch, R.; Young, R.A. Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell Stem Cell 2010, 7, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Maherali, N.; Sridharan, R.; Xie, W.; Utikal, J.; Eminli, S.; Arnold, K.; Stadtfeld, M.; Yachechko, R.; Tchieu, J.; Jaenisch, R.; et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 2007, 1, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Hanna, J.; Zhang, X.; Ku, M.; Wernig, M.; Schorderet, P.; Bernstein, B.E.; Jaenisch, R.; Lander, E.S.; Meissner, A. Dissecting direct reprogramming through integrative genomic analysis. Nature 2008, 454, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Schoenfelder, S.; Sugar, R.; Dimond, A.; Javierre, B.-M.; Armstrong, H.; Mifsud, B.; Dimitrova, E.; Matheson, L.; Tavares-Cadete, F.; Furlan-Magaril, M.; et al. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat. Genet. 2015, 47, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Beagrie, R.A.; Pombo, A. Gene activation by metazoan enhancers: Diverse mechanisms stimulate distinct steps of transcription. Bioessays 2016, 38, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Yu, N.-K.; Kaang, B.-K. CTCF as a multifunctional protein in genome regulation and gene expression. Exp. Mol. Med. 2015, 47, e166. [Google Scholar] [CrossRef] [PubMed]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: what, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Ji, F.; Sunwoo, H.; Jain, G.; Lee, J.T.; Sadreyev, R.I.; Dekker, J.; Kingston, R.E. Polycomb Repressive Complex 1 Generates Discrete Compacted Domains that Change during Differentiation. Mol. Cell 2017, 65, 432–446.e5. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.D.; Sansom, S.N.; Smith, J.; Dobenecker, M.-W.; Tarakhovsky, A.; Livesey, F.J. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 15957–15962. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, Y.; Suzki, N.; Tsuboi, M.; Endo, T.A.; Toyoda, T.; Shinga, J.; Koseki, H.; Vidal, M.; Gotoh, Y. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 2009, 63, 600–613. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, F. Spatial and temporal specification of neural fates by transcription factor codes. Development 2007, 134, 3771–3780. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ge, W.; Martinowich, K.; Becker-Catania, S.; Coskun, V.; Zhu, W.; Wu, H.; Castro, D.; Guillemot, F.; Fan, G.; et al. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat. Neurosci. 2005, 8, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Petryniak, M.A.; Potter, G.B.; Rowitch, D.H.; Rubenstein, J.L. R. Dlx1 and Dlx2 control neuronal versus oligodendroglial cell fate acquisition in the developing forebrain. Neuron 2007, 55, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Ghavi-Helm, Y.; Klein, F.A.; Pakozdi, T.; Ciglar, L.; Noordermeer, D.; Huber, W.; Furlong, E.E.M. Enhancer loops appear stable during development and are associated with paused polymerase. Nature 2014, 512, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Lonfat, N.; Montavon, T.; Darbellay, F.; Gitto, S.; Duboule, D. Convergent evolution of complex regulatory landscapes and pleiotropy at Hox loci. Science 2014, 346, 1004–1006. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.W.; Salinas, R.D.; Siu, J.J.; Kelley, K.W.; Delgado, R.N.; Paredes, M.F.; Alvarez-Buylla, A.; Oldham, M.C.; Lim, D.A. Distinct and separable roles for EZH2 in neurogenic astroglia. eLife 2014, 3, e02439. [Google Scholar] [CrossRef] [PubMed]
- Román-Trufero, M.; Méndez-Gómez, H.R.; Pérez, C.; Hijikata, A.; Fujimura, Y.; Endo, T.; Koseki, H.; Vicario-Abejón, C.; Vidal, M. Maintenance of undifferentiated state and self-renewal of embryonic neural stem cells by Polycomb protein Ring1B. Stem Cells 2009, 27, 1559–1570. [Google Scholar] [CrossRef] [PubMed]
- Van der Lugt, N.M.; Domen, J.; Linders, K.; van Roon, M.; Robanus-Maandag, E.; te Riele, H.; van der Valk, M.; Deschamps, J.; Sofroniew, M.; van Lohuizen, M. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 1994, 8, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Pardal, R.; Iwashita, T.; Park, I.-K.; Clarke, M.F.; Morrison, S.J. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 2003, 425, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Lingbeek, M.; Shakhova, O.; Liu, J.; Tanger, E.; Saremaslani, P.; Van Lohuizen, M.; Marino, S. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature 2004, 428, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.; Scheijen, B.; Voncken, J.W.; Kieboom, K.; Berns, A.; van Lohuizen, M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999, 13, 2678–2690. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, S.W.M.; Valk-Lingbeek, M.E.; van der Stoop, P.P.M.; Jacobs, J.J.L.; Kieboom, K.; Tanger, E.; Hulsman, D.; Leung, C.; Arsenijevic, Y.; Marino, S.; et al. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev. 2005, 19, 1438–1443. [Google Scholar] [CrossRef] [PubMed]
- Fasano, C.A.; Phoenix, T.N.; Kokovay, E.; Lowry, N.; Elkabetz, Y.; Dimos, J.T.; Lemischka, I.R.; Studer, L.; Temple, S. Bmi-1 cooperates with Foxg1 to maintain neural stem cell self-renewal in the forebrain. Genes Dev. 2009, 23, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Wang, Y.; Dimos, J.T.; Fasano, C.A.; Phoenix, T.N.; Lemischka, I.R.; Ivanova, N.B.; Stifani, S.; Morrisey, E.E.; Temple, S. The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat. Neurosci. 2006, 9, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Tümpel, S.; Wiedemann, L.M.; Krumlauf, R. Hox genes and segmentation of the vertebrate hindbrain. Curr. Top. Dev. Biol. 2009, 88, 103–137. [Google Scholar] [CrossRef] [PubMed]
- Di Meglio, T.; Kratochwil, C.F.; Vilain, N.; Loche, A.; Vitobello, A.; Yonehara, K.; Hrycaj, S.M.; Roska, B.; Peters, A.H.F.M.; Eichmann, A.; et al. Ezh2 orchestrates topographic migration and connectivity of mouse precerebellar neurons. Science 2013, 339, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Burgold, T.; Voituron, N.; Caganova, M.; Tripathi, P.P.; Menuet, C.; Tusi, B.K.; Spreafico, F.; Bévengut, M.; Gestreau, C.; Buontempo, S.; et al. The H3K27 demethylase JMJD3 is required for maintenance of the embryonic respiratory neuronal network, neonatal breathing, and survival. Cell Rep. 2012, 2, 1244–1258. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Liu, S.; Qin, R.; Zhang, Y.; Wang, G.; Shang, Y.; Wang, Y.; Liang, J. Coordinated regulation of dendrite arborization by epigenetic factors CDYL and EZH. J. Neurosci. 2014, 34, 4494–4508. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Sawada, J.; Sui, G.; Affar, E.B.; Whetstine, J.R.; Lan, F.; Ogawa, H.; Luke, M.P.-S.; Nakatani, Y.; Shi, Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 2003, 422, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, M.; Eberl, H.C.; Matarese, F.; Marks, H.; Denissov, S.; Butter, F.; Lee, K.K.; Olsen, J.V.; Hyman, A.A.; Stunnenberg, H.G.; et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 2010, 142, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, X.; Gui, B.; Xie, G.; Zhang, D.; Shang, Y.; Liang, J. Corepressor protein CDYL functions as a molecular bridge between polycomb repressor complex 2 and repressive chromatin mark trimethylated histone lysine 27. J. Biol. Chem. 2011, 286, 42414–42425. [Google Scholar] [CrossRef] [PubMed]
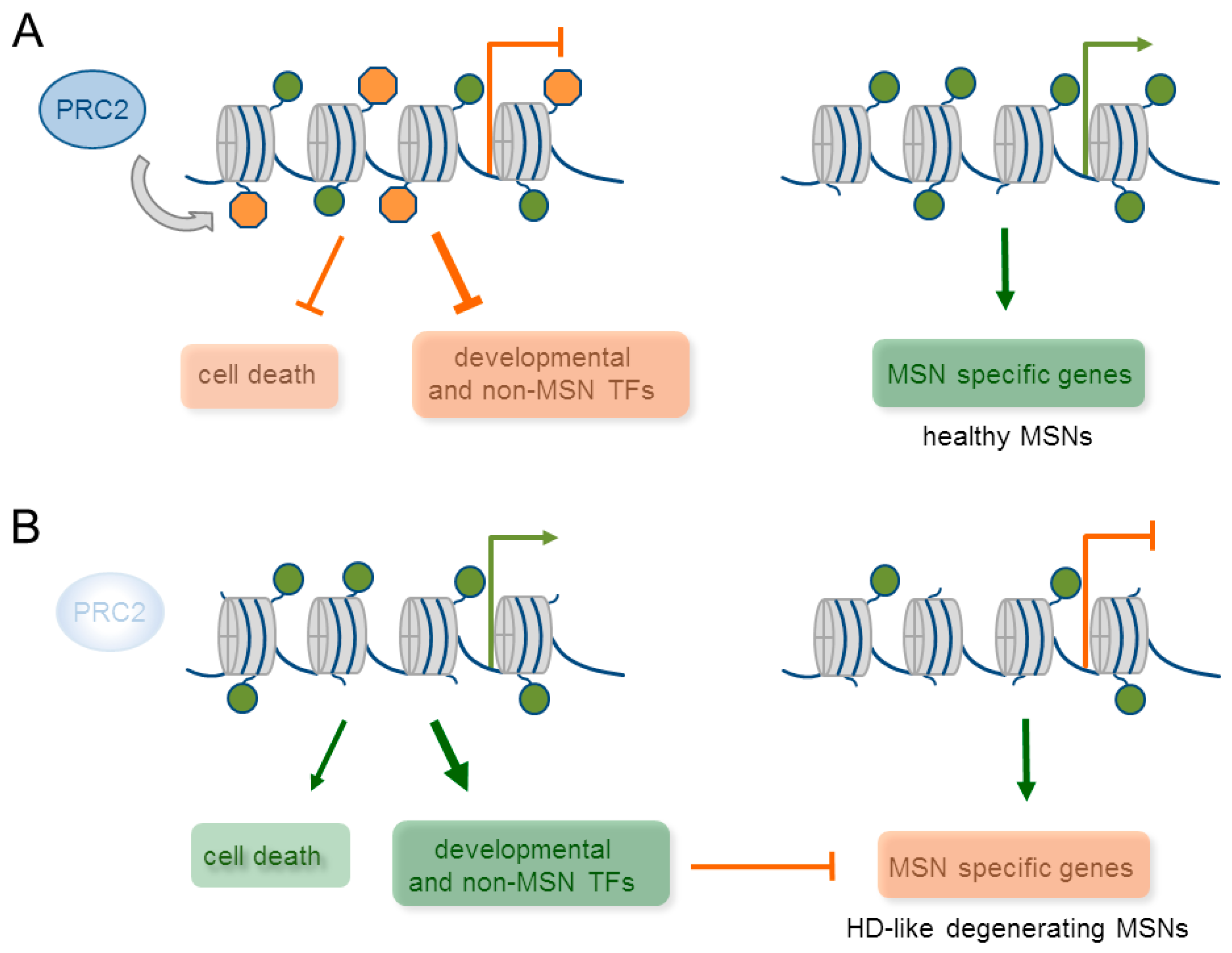
- Von Schimmelmann, M.; Feinberg, P.A.; Sullivan, J.M.; Ku, S.M.; Badimon, A.; Duff, M.K.; Wang, Z.; Lachmann, A.; Dewell, S.; Ma’ayan, A.; et al. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci. 2016, 19, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- He, X.-B.; Kim, M.; Kim, S.-Y.; Yi, S.-H.; Rhee, Y.-H.; Kim, T.; Lee, E.-H.; Park, C.-H.; Dixit, S.; Harrison, F.E.; et al. Vitamin C facilitates dopamine neuron differentiation in fetal midbrain through TET1- and JMJD3-dependent epigenetic control manner. Stem Cells 2015, 33, 1320–1332. [Google Scholar] [CrossRef] [PubMed]
- Palomer, E.; Carretero, J.; Benvegnù, S.; Dotti, C.G.; Martin, M.G. Neuronal activity controls Bdnf expression via Polycomb de-repression and CREB/CBP/JMJD3 activation in mature neurons. Nat. Commun. 2016, 7, 11081. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hart, R.P.; Mallimo, E.M.; Swerdel, M.R.; Kusnecov, A.W.; Herrup, K. EZH2-mediated H3K27 trimethylation mediates neurodegeneration in ataxia-telangiectasia. Nat. Neurosci. 2013, 16, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Södersten, E.; Feyder, M.; Lerdrup, M.; Gomes, A.-L.; Kryh, H.; Spigolon, G.; Caboche, J.; Fisone, G.; Hansen, K. Dopamine signaling leads to loss of Polycomb repression and aberrant gene activation in experimental parkinsonism. PLoS Genet. 2014, 10, e1004574. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Tsuji, J.; Labadorf, A.; Roussos, P.; Chen, J.-F.; Myers, R.H.; Akbarian, S.; Weng, Z. The Role of H3K4me3 in Transcriptional Regulation Is Altered in Huntington’s Disease. PLoS ONE 2015, 10, e0144398. [Google Scholar] [CrossRef] [PubMed]
- Seong, I.S.; Woda, J.M.; Song, J.-J.; Lloret, A.; Abeyrathne, P.D.; Woo, C.J.; Gregory, G.; Lee, J.-M.; Wheeler, V.C.; Walz, T.; et al. Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 2010, 19, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Biagioli, M.; Ferrari, F.; Mendenhall, E.M.; Zhang, Y.; Erdin, S.; Vijayvargia, R.; Vallabh, S.M.; Solomos, N.; Manavalan, P.; Ragavendran, A.; et al. Htt CAG repeat expansion confers pleiotropic gains of mutant huntingtin function in chromatin regulation. Hum. Mol. Genet. 2015, 24, 2442–2457. [Google Scholar] [CrossRef] [PubMed]
- Hoss, A.G.; Kartha, V.K.; Dong, X.; Latourelle, J.C.; Dumitriu, A.; Hadzi, T.C.; Macdonald, M.E.; Gusella, J.F.; Akbarian, S.; Chen, J.-F.; et al. MicroRNAs located in the Hox gene clusters are implicated in huntington’s disease pathogenesis. PLoS Genet. 2014, 10, e1004188. [Google Scholar] [CrossRef] [PubMed]
- Labadorf, A.; Hoss, A.G.; Lagomarsino, V.; Latourelle, J.C.; Hadzi, T.C.; Bregu, J.; MacDonald, M.E.; Gusella, J.F.; Chen, J.-F.; Akbarian, S.; et al. RNA Sequence Analysis of Human Huntington Disease Brain Reveals an Extensive Increase in Inflammatory and Developmental Gene Expression. PLoS ONE 2015, 10, e0143563. [Google Scholar] [CrossRef] [PubMed]
- Gaspard, N.; Vanderhaeghen, P. Mechanisms of neural specification from embryonic stem cells. Curr. Opin. Neurobiol. 2010, 20, 37–43. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, A.; Sportelli, V.; Ziller, M.; Spengler, D. Switch-Like Roles for Polycomb Proteins from Neurodevelopment to Neurodegeneration. Epigenomes 2017, 1, 21. https://doi.org/10.3390/epigenomes1030021
Hoffmann A, Sportelli V, Ziller M, Spengler D. Switch-Like Roles for Polycomb Proteins from Neurodevelopment to Neurodegeneration. Epigenomes. 2017; 1(3):21. https://doi.org/10.3390/epigenomes1030021
Chicago/Turabian StyleHoffmann, Anke, Vincenza Sportelli, Michael Ziller, and Dietmar Spengler. 2017. "Switch-Like Roles for Polycomb Proteins from Neurodevelopment to Neurodegeneration" Epigenomes 1, no. 3: 21. https://doi.org/10.3390/epigenomes1030021
APA StyleHoffmann, A., Sportelli, V., Ziller, M., & Spengler, D. (2017). Switch-Like Roles for Polycomb Proteins from Neurodevelopment to Neurodegeneration. Epigenomes, 1(3), 21. https://doi.org/10.3390/epigenomes1030021