
PRC1 Prevents Replication Stress during Chondrogenic Transit Amplification

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
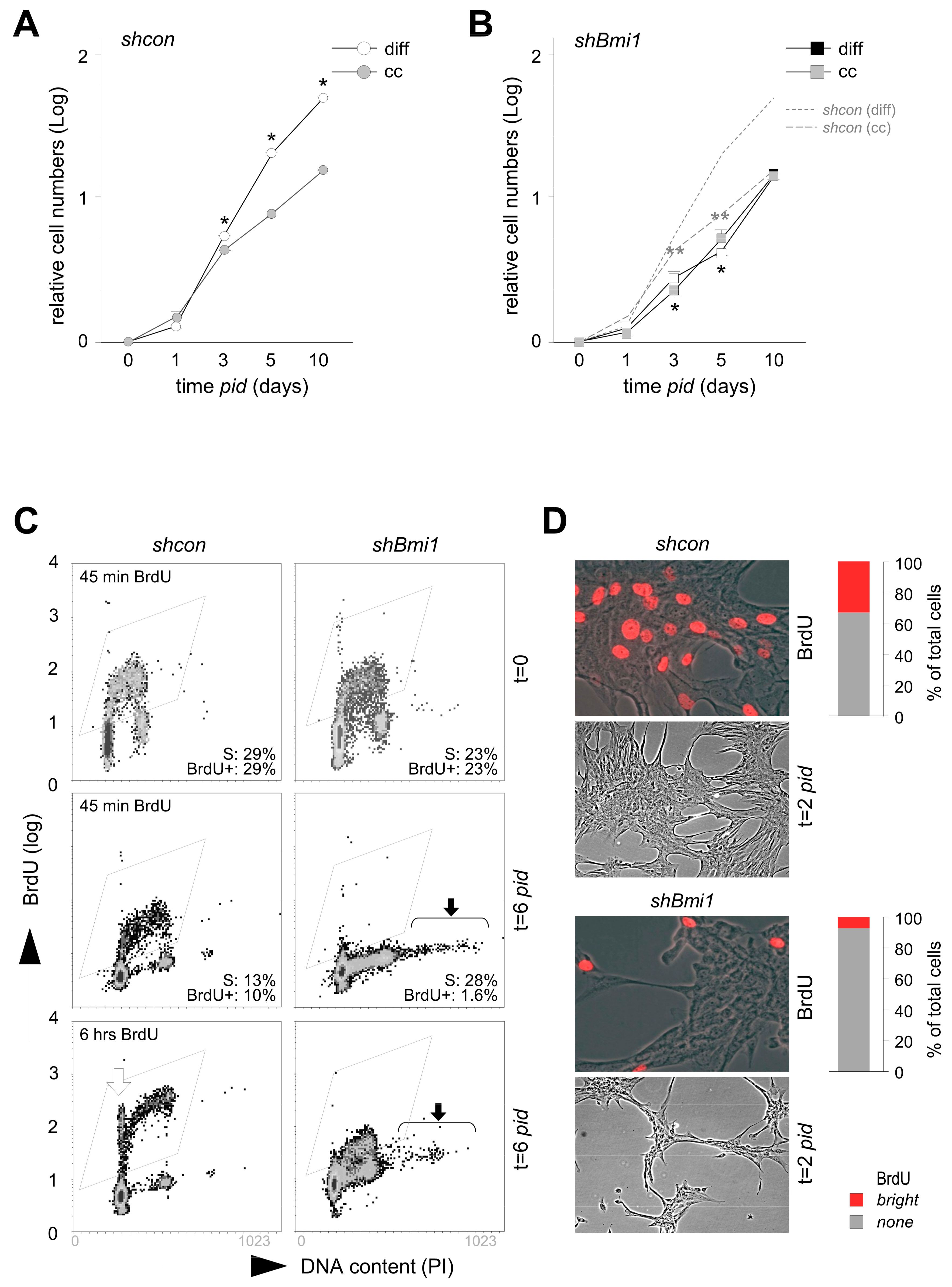
2.1. Loss of PRC1 Prevents Transit Amplification and Reduces Chondrogenic Capacity
2.2. BMI1-Deficiency Leads to Increased Replication-Associated DNA Damage during Transit Amplification
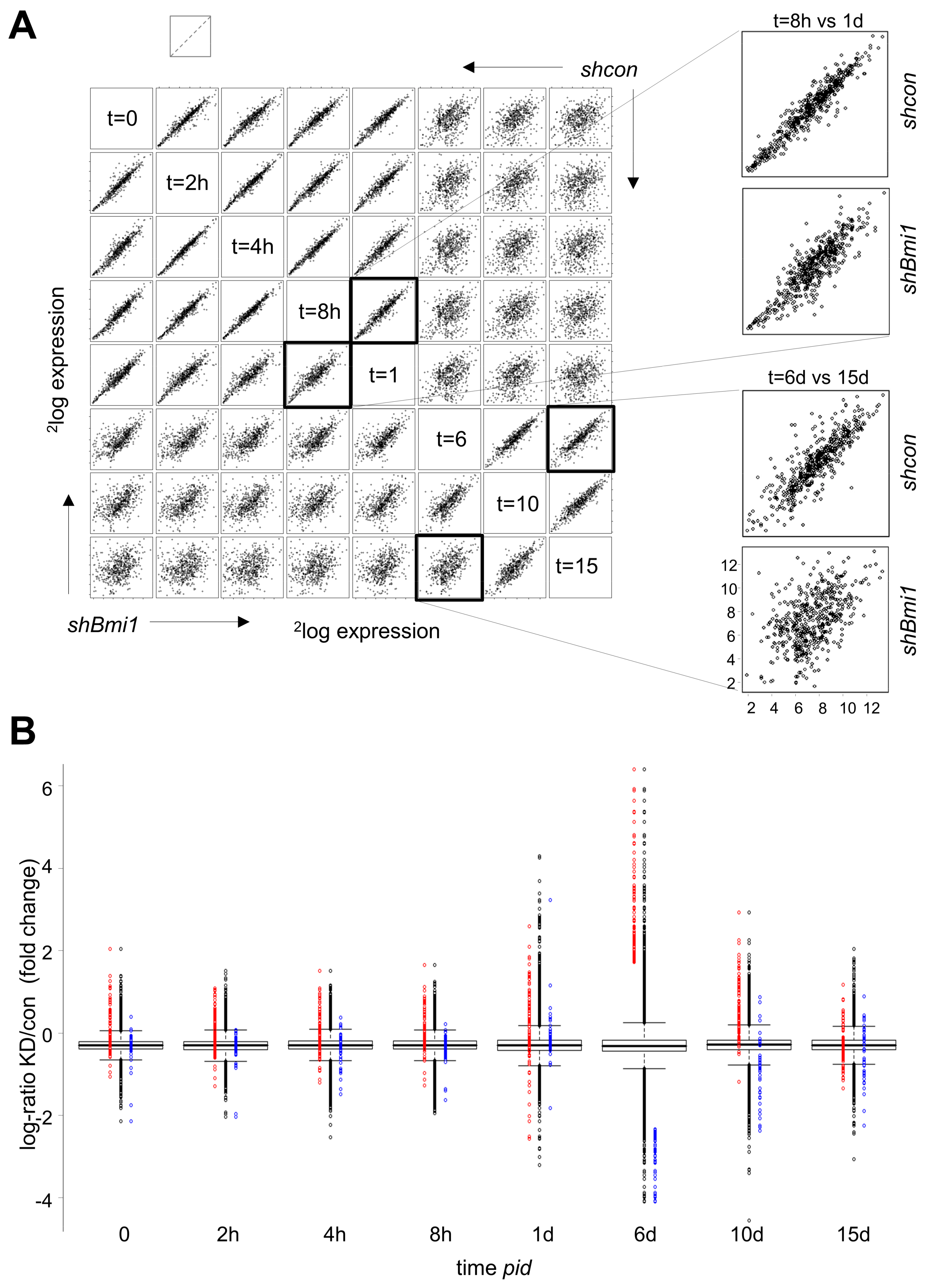
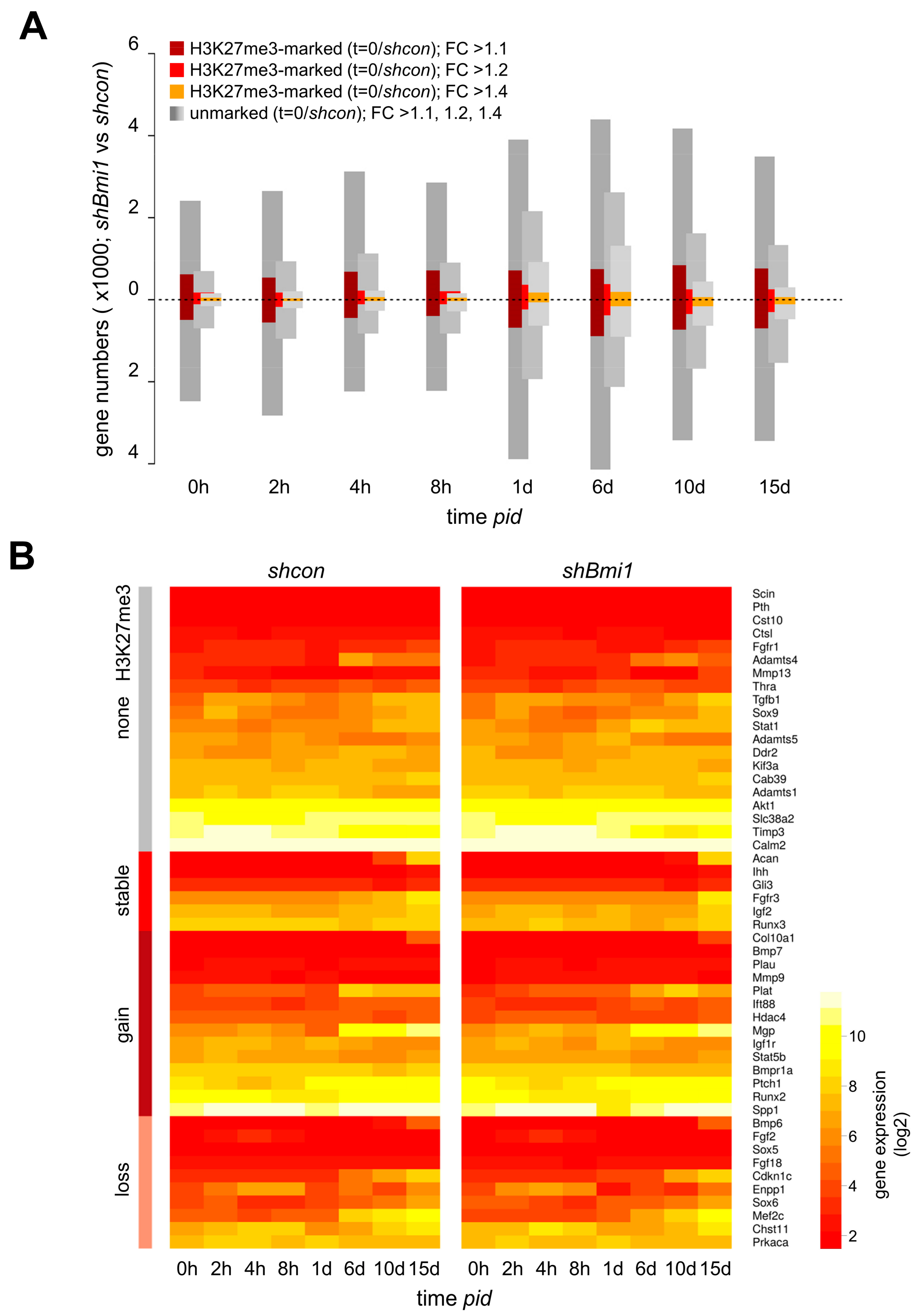
2.3. H3K27me3 Is not a Major Determinant of Transcriptional Repression during Differentiation
2.4. Abnormal TOP2A Cleavable-Complex Processing in PRC1-Deficient Cells Causes Double Strand DNA Breaks
2.5. Increased POLR2 Phosphorylation and Stalled Transcription Forks in BMI1-Depleted Cells
2.6. BMI1 Prevents Premature Replication-Associated Senescence during Chondrogenesis
3. Discussion
4. Methods
4.1. Cell Culture, Differentiation Assays
4.2. Retroviral Vectors, Infections
- shBmi1[m + h] (ENSMUSG00000028796): 5′-GTTCACAAGACCAGACCAC-3′,
- shRnf2[m] (ENSMUSG00000028796): 5′-GTATCTGGCTGTGAGGTTA-3′,
- shPhc2[m] (ENSMUSG00000028796): 5′-GTTCAAGCGTTCCAAGCGC-3′.
4.3. Proliferation Assays
4.4. Immunoblot Analysis
4.5. Immunofluorescence
4.6. Antibodies
4.7. Single Cell Gel Electrophoresis Assay (Neutral Comet Assay)
4.8. RNA Isolation, cDNA Synthesis, Quantitative PCR Analysis
4.9. Affymetrix Expression Arrays, Pathway Analysis
4.10. H3k27me3 Chip-Seq, Data Processing: Alignment, Normalization, Background Correction, Identification of Enriched Regions
4.11. H3K27me3-Enrichment Data Summarization, Integration Gene Expression Data
4.12. Cross-Link Immunoprecipitation (X-IP), Deep-Sequencing, Chromatin-Association Studies
4.13. Senescence-Associated Beta-Galactosidase Staining
4.14. Mice, Tissue Histology
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BMI1 | B lymphoma Mo-MLV insertion region 1 |
| BrdU | bromo-deoxyuridine |
| ChIP | chomatin immunoprecipitation |
| DDR | DNA damage response |
| DPOL2 | DNA polymerase 2 |
| DPOLR2 | RNA polymerase 2 |
| DSB | double strand DNA breaks |
| ERSS | ER-stress signaling |
| GMNN | geminin |
| H3K27me3 | histon H3 Lysine 27 trimethylation |
| H3K9me3 | histon H3 Lysine 9 trimethylation |
| HZ | hypertrophic zone |
| IF | immunofluorescence |
| KD | knockdown |
| KMD | Lysine demethylase |
| KMT | Lysine methyl transferase |
| OE | overexpression |
| OIS | oncogene induced senescence |
| PHC2 | Polyhomeotic 2 |
| pid | post-induction of differentiation |
| PRC | Polycomb repressive complex |
| pre-HZ | pre-hypertrophic zone |
| PZ | proliferative zone |
| RING1 | really interesting new gene 1A |
| RNF2 | ring-finger protein 2 |
| SA-bGAL | senescence associated beta-galactosidase |
| SASP | senescence-associated Secretory phenotype |
| shcon | control shRNA |
| shRNA | short hairpin RNA |
| SLR | senescence like response |
| SMS | senescence messaging secretome |
| TA | transit amplification |
| TOP2A | topoisomerase 2 |
| TrxG | Trithorax group proteins |
| UPR | unfolded protein response |
| yH2A.X | H2A.X Serine 139 phosphorylation |
References
- Prezioso, C.; Orlando, V. Polycomb proteins in mammalian cell differentiation and plasticity. FEBS Lett. 2011, 585, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Verrijzer, P. Biochemical mechanisms of gene regulation by polycomb group protein complexes. Curr. Opin. Genet. Dev. 2009, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A. Chromatin compaction at Hox loci: A polycomb tale beyond histone tails. Mol. Cell 2010, 38, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, J.; van den Akker, E.; Forlani, S.; De Graaff, W.; Oosterveen, T.; Roelen, B.; Roelfsema, J. Initiation, establishment and maintenance of Hox gene expression patterns in the mouse. Int. J. Dev. Biol. 1999, 43, 635–650. [Google Scholar] [PubMed]
- Scelfo, A.; Piunti, A.; Pasini, D. The controversial role of the polycomb group proteins in transcription and cancer: How much do we not understand polycomb proteins? FEBS J. 2015, 282, 1703–1722. [Google Scholar] [CrossRef] [PubMed]
- Van Lohuizen, M. Functional analysis of mouse polycomb group genes. Cell. Mol. Life Sci. 1998, 54, 71–79. [Google Scholar] [CrossRef] [PubMed]
- O’Dor, E.; Beck, S.A.; Brock, H.W. Polycomb group mutants exhibit mitotic defects in syncytial cell cycles of drosophila embryos. Dev. Biol. 2005. [Google Scholar] [CrossRef] [PubMed]
- Soshnikova, N.; Duboule, D. Epigenetic regulation of vertebrate Hox genes: A dynamic equilibrium. Epigenetics 2009, 4, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.M.; Cavalli, G. The role of polycomb group proteins in cell cycle regulation during development. Cell Cycle 2006, 5, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Sparmann, A.; van Lohuizen, M. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 2006, 6, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Ismail, I.H.; Andrin, C.; McDonald, D.; Hendzel, M.J. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J. Cell Biol. 2010, 191, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Gieni, R.S.; Ismail, I.H.; Campbell, S.; Hendzel, M.J. Polycomb group proteins in the DNA damage response: A link between radiation resistance and “stemness”. Cell Cycle 2011, 10, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Voncken, J.W.; Roelen, B.A.; Roefs, M.; de Vries, S.; Verhoeven, E.; Marino, S.; Deschamps, J.; van Lohuizen, M. Rnf2 (RING1b) deficiency causes gastrulation arrest and cell cycle inhibition. Proc. Natl. Acad. Sci. USA 2003, 100, 2468–2473. [Google Scholar] [CrossRef] [PubMed]
- Lessard, J.; Sauvageau, G. BMI-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003, 423, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; He, S.; Bydon, M.; Morrison, S.J.; Pardal, R. BMI-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16ink4a and p19arf senescence pathways. Genes Dev. 2005, 19, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Morrison, S.J.; Clarke, M.F. BMI1, stem cells and senescence regulation. J. Clin. Investig. 2004, 113, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Lapthanasupkul, P.; Feng, J.; Mantesso, A.; Takada-Horisawa, Y.; Vidal, M.; Koseki, H.; Wang, L.; An, Z.; Miletich, I.; Sharpe, P.T. RING1a/b polycomb proteins regulate the mesenchymal stem cell niche in continuously growing incisors. Dev. Biol. 2012, 367, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Itzkovitz, S.; Lyubimova, A.; Blat, I.C.; Maynard, M.; van Es, J.; Lees, J.; Jacks, T.; Clevers, H.; van Oudenaarden, A. Single-molecule transcript counting of stem-cell markers in the mouse intestine. Nat. Cell Biol. 2012, 14, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaapen, F.; van den Akker, G.G.; Caron, M.M.; Prickaerts, P.; Rofel, C.; Dahlmans, V.E.; Surtel, D.A.; Paulis, Y.; Schweizer, F.; Welting, T.J.; et al. The immediate early gene product EGR1 and polycomb group proteins interact in epigenetic programming during chondrogenesis. PLoS ONE 2013, 8, e58083. [Google Scholar] [CrossRef] [PubMed]
- Mackie, E.J.; Ahmed, Y.A.; Tatarczuch, L.; Chen, K.S.; Mirams, M. Endochondral ossification: How cartilage is converted into bone in the developing skeleton. Int. J. Biochem. Cell Biol. 2008, 40, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, T.; Miwa, Y.; Kimata, K.; Ikawa, Y. A chondrogenic cell line derived from a differentiating culture of AT805 teratocarcinoma cells. Cell Differ. Dev. 1990, 30, 109–116. [Google Scholar] [CrossRef]
- Suzuki, M.; Mizutani-Koseki, Y.; Fujimura, Y.; Miyagishima, H.; Kaneko, T.; Takada, Y.; Akasaka, T.; Tanzawa, H.; Takihara, Y.; Nakano, M.; et al. Involvement of the polycomb-group gene RING1b in the specification of the anterior-posterior axis in mice. Development 2002, 129, 4171–4183. [Google Scholar] [PubMed]
- Van der Lugt, N.M.; Domen, J.; Linders, K.; van Roon, M.; Robanus-Maandag, E.; te Riele, H.; van der Valk, M.; Deschamps, J.; Sofroniew, M.; van Lohuizen, M.; et al. Posterior transformation, neurological abnormalities and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 1994, 8, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Isono, K.; Fujimura, Y.; Shinga, J.; Yamaki, M.; O-Wang, J.; Takihara, Y.; Murahashi, Y.; Takada, Y.; Mizutani-Koseki, Y.; Koseki, H. Mammalian polyhomeotic homologues Phc2 and Phc1 act in synergy to mediate polycomb repression of Hox genes. Mol. Cell. Biol. 2005, 25, 6694–6706. [Google Scholar] [CrossRef] [PubMed]
- Bel, S.; Core, N.; Djabali, M.; Kieboom, K.; Van der, L.N.; Alkema, M.J.; van Lohuizen, M. Genetic interactions and dosage effects of polycomb group genes in mice. Development 1998, 125, 3543–3551. [Google Scholar] [PubMed]
- Del Mar, L.M.; Marcos-Gutierrez, C.; Perez, C.; Schoorlemmer, J.; Ramirez, A.; Magin, T.; Vidal, M. Loss- and gain-of-function mutations show a polycomb group function for ring1a in mice. Development 2000, 127, 5093–5100. [Google Scholar]
- Celis, J.E.; Celis, A. Cell cycle-dependent variations in the distribution of the nuclear protein cyclin proliferating cell nuclear antigen in cultured cells: Subdivision of s phase. Proc. Natl. Acad. Sci. USA 1985, 82, 3262–3266. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fumagalli, M.; d’Adda di Fagagna, F. Breaking news: High-speed race ends in arrest—How oncogenes induce senescence. Trends Cell Biol. 2007, 17, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Redon, C.; Rao, V.A.; Seiler, J.A.; Sordet, O.; Takemura, H.; Antony, S.; Meng, L.; Liao, Z.; Kohlhagen, G.; et al. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2003, 532, 173–203. [Google Scholar] [CrossRef]
- Fisher, C.L.; Fisher, A.G. Chromatin states in pluripotent, differentiated and reprogrammed cells. Curr. Opin. Genet. Dev. 2011, 21, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Surface, L.E.; Thornton, S.R.; Boyer, L.A. Polycomb group proteins set the stage for early lineage commitment. Cell Stem Cell 2010, 7, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Prickaerts, P.; Adriaens, M.E.; Beucken, T.V.D.; Koch, E.; Dubois, L.; Dahlmans, V.E.H.; Gits, C.; Evelo, C.T.A.; Chan-Seng-Yue, M.; Wouters, B.G.; et al. Hypoxia increases genome-wide bivalent epigenetic marking by specific gain of H3K27me3. Epigenetics Chromatin 2016, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Roca, J. Topoisomerase II: A fitted mechanism for the chromatin landscape. Nucleic Acids Res. 2009, 37, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Leppard, J.B.; Champoux, J.J. Human DNA topoisomerase I: Relaxation, roles and damage control. Chromosoma 2005, 114, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Petruk, S.; Sedkov, Y.; Johnston, D.M.; Hodgson, J.W.; Black, K.L.; Kovermann, S.K.; Beck, S.; Canaani, E.; Brock, H.W.; Mazo, A. TrxG and PcG proteins but not methylated histones remain associated with DNA through replication. Cell 2012, 150, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Lupo, R.; Breiling, A.; Bianchi, M.E.; Orlando, V. Drosophila chromosome condensation proteins topoisomerase II and barren colocalize with polycomb and maintain Fab-7 PRE silencing. Mol. Cell 2001, 7, 127–136. [Google Scholar] [CrossRef]
- Piunti, A.; Rossi, A.; Cerutti, A.; Albert, M.; Jammula, S.; Scelfo, A.; Cedrone, L.; Fragola, G.; Olsson, L.; Koseki, H.; et al. Polycomb proteins control proliferation and transformation independently of cell cycle checkpoints by regulating DNA replication. Nat. Commun. 2014, 5, 3649. [Google Scholar] [CrossRef] [PubMed]
- Alchanati, I.; Teicher, C.; Cohen, G.; Shemesh, V.; Barr, H.M.; Nakache, P.; Ben-Avraham, D.; Idelevich, A.; Angel, I.; Livnah, N.; et al. The E3 ubiquitin-ligase Bmi1/Ring1A controls the proteasomal degradation of top2α cleavage complex—A potentially new drug target. PLoS ONE 2009, 4, e8104. [Google Scholar] [CrossRef] [PubMed]
- Deweese, J.E.; Osheroff, N. The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing. Nucleic Acids Res. 2009, 37, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, L.; Ferrari, R.; Vashisht, A.A.; Wohlschlegel, J.A.; Kurdistani, S.K.; Carey, M. Polycomb repressive complex 1 (PRC1) disassembles RNA polymerase II preinitiation complexes. J. Biol. Chem. 2012, 287, 35784–35794. [Google Scholar] [CrossRef] [PubMed]
- Hsin, J.P.; Manley, J.L. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012, 26, 2119–2137. [Google Scholar] [CrossRef] [PubMed]
- Saxowsky, T.T.; Doetsch, P.W. RNA polymerase encounters with DNA damage: Transcription-coupled repair or transcriptional mutagenesis? Chem. Rev. 2006, 106, 474–488. [Google Scholar] [CrossRef] [PubMed]
- Sordet, O.; Larochelle, S.; Nicolas, E.; Stevens, E.V.; Zhang, C.; Shokat, K.M.; Fisher, R.P.; Pommier, Y. Hyperphosphorylation of RNA polymerase II in response to topoisomerase I cleavage complexes and its association with transcription- and BRCA1-dependent degradation of topoisomerase I. J. Mol. Biol. 2008, 381, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.W.; Hummert, P.; Mills, J.C.; Kroll, K.L. Geminin cooperates with polycomb to restrain multi-lineage commitment in the early embryo. Development 2010, 138, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, M.; Yasunaga, S.; Ohno, Y.; Tsumura, M.; Okada, S.; Ishikawa, N.; Shirao, K.; Kikuchi, A.; Nishitani, H.; Kobayashi, M.; et al. Polycomb-group complex 1 acts as an E3 ubiquitin ligase for geminin to sustain hematopoietic stem cell activity. Proc. Natl. Acad. Sci. USA 2008, 105, 10396–10401. [Google Scholar] [CrossRef] [PubMed]
- Melixetian, M.; Helin, K. Geminin: A major DNA replication safeguard in higher eukaryotes. Cell Cycle 2004, 3, 1002–1004. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Yang, X.; Takihara, Y.; Knoetgen, H.; Kessel, M. The cell-cycle regulator geminin inhibits Hox function through direct and polycomb-mediated interactions. Nature 2004, 427, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Narita, M. Cellular senescence and chromatin organisation. Br. J. Cancer 2007, 96, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Fasano, C.A.; Dimos, J.T.; Ivanova, N.B.; Lowry, N.; Lemischka, I.R.; Temple, S. shRNA knockdown of Bmi-1 reveals a critical role for p21-Rb pathway in NSC self-renewal during development. Cell Stem Cell 2007, 1, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Negishi, M.; Saraya, A.; Miyagi, S.; Nagao, K.; Inagaki, Y.; Nishikawa, M.; Tajima, S.; Koseki, H.; Tsuda, H.; Takasaki, Y.; et al. BMI1 cooperates with Dnmt1-associated protein 1 in gene silencing. Biochem. Biophys. Res. Commun. 2007, 353, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Prickaerts, P.; Niessen, H.E.C.; Mouchel-Vielh, E.; Dahlmans, V.E.H.; van den Akker, G.G.H.; Geijselaers, C.; Adriaens, M.E.; Spaapen, F.; Takihara, Y.; Rapp, U.R.; et al. MK3 controls polycomb target gene expression via negative feedback on ERK. Epigenetics Chromatin 2012, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Vanhove, J.; Pistoni, M.; Welters, M.; Eggermont, K.; Vanslembrouck, V.; Helsen, N.; Boon, R.; Najimi, M.; Sokal, E.; Collas, P.; et al. H3K27me3 does not orchestrate the expression of lineage-specific markers in hesc-derived hepatocytes in vitro. Stem Cell Rep. 2016, 7, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Adriaens, M.E.; Prickaerts, P.; Chan-Seng-Yue, M.; van den Beucken, T.; Dahlmans, V.E.H.; Eijssen, L.M.; Beck, T.; Wouters, B.G.; Voncken, J.W.; Evelo, C.T.A. Quantitative analysis of ChIP-seq data uncovers dynamic and sustained H3K4me3 and H3K27me3 modulation in cancer cells under hypoxia. Epigenetics Chromatin 2016, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Morey, L.; Santanach, A.; Blanco, E.; Aloia, L.; Nora, E.P.; Bruneau, B.G.; Di Croce, L. Polycomb regulates mesoderm cell fate-specification in embryonic stem cells through activation and repression mechanisms. Cell Stem Cell 2015, 17, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Lee, P.; Stafford, J.M.; von Schimmelmann, M.; Schaefer, A.; Reinberg, D. An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 2014, 516, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, C.A.; Misulovin, Z.; Gause, M.; Koenig, A.; Gohara, D.W.; Watson, A.; Dorsett, D. Cohesin and polycomb proteins functionally interact to control transcription at silenced and active genes. PLoS Genet. 2013, 9, e1003560. [Google Scholar] [CrossRef] [PubMed]
- Frangini, A.; Sjoberg, M.; Roman-Trufero, M.; Dharmalingam, G.; Haberle, V.; Bartke, T.; Lenhard, B.; Malumbres, M.; Vidal, M.; Dillon, N. The aurora B kinase and the polycomb protein RING1B combine to regulate active promoters in quiescent lymphocytes. Mol. Cell 2013, 51, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Creppe, C.; Palau, A.; Malinverni, R.; Valero, V.; Buschbeck, M. A cbx8-containing polycomb complex facilitates the transition to gene activation during ES cell differentiation. PLoS Genet. 2014, 10, e1004851. [Google Scholar] [CrossRef] [PubMed]
- Fragola, G.; Germain, P.L.; Laise, P.; Cuomo, A.; Blasimme, A.; Gross, F.; Signaroldi, E.; Bucci, G.; Sommer, C.; Pruneri, G.; et al. Cell reprogramming requires silencing of a core subset of polycomb targets. PLoS Genet. 2013, 9, e1003292. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.; Tursun, B.; Rahe, D.P.; Hobert, O. Removal of polycomb repressive complex 2 makes C. Elegans germ cells susceptible to direct conversion into specific somatic cell types. Cell Rep. 2012, 2, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Hemberger, M.; Dean, W.; Reik, W. Epigenetic dynamics of stem cells and cell lineage commitment: Digging waddington’s canal. Nat. Rev. Mol. Cell Biol. 2009, 10, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xu, L.; Xu, Q.; Li, D.; Yang, Y.; Karsenty, G.; Chen, C.D. JMJD3 promotes chondrocyte proliferation and hypertrophy during endochondral bone formation in mice. J. Mol. Cell Biol. 2015, 7, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Fullgrabe, J.; Hajji, N.; Joseph, B. Cracking the death code: Apoptosis-related histone modifications. Cell Death Differ. 2010, 17, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Basenko, E.Y.; Sasaki, T.; Ji, L.; Prybol, C.J.; Burckhardt, R.M.; Schmitz, R.J.; Lewis, Z.A. Genome-wide redistribution of H3K27me3 is linked to genotoxic stress and defective growth. Proc. Natl. Acad. Sci. USA 2015, 112, E6339–E6348. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.H.; Bracken, A.P.; Pasini, D.; Dietrich, N.; Gehani, S.S.; Monrad, A.; Rappsilber, J.; Lerdrup, M.; Helin, K. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 2008, 10, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Samarabandu, J.; Devdhar, R.S.; Siegel, A.J.; Acharya, R.; Berezney, R. Segregation of transcription and replication sites into higher order domains. Science 1998, 281, 1502–1506. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. The checkpoint response to replication stress. DNA Repair (Amst.) 2009, 8, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, C.J.; Dhillon, P.; Moore, T.; Lloyd, R.G. Avoiding and resolving conflicts between DNA replication and transcription. DNA Repair (Amst.) 2007, 6, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Voncken, J.W.; Schweizer, D.; Aagaard, L.; Sattler, L.; Jantsch, M.F.; van Lohuizen, M. Chromatin-association of the polycomb group protein BMI1 is cell cycle-regulated and correlates with its phosphorylation status. J. Cell Sci. 1999, 112 (Pt 24), 4627–4639. [Google Scholar] [PubMed]
- Francis, N.J.; Follmer, N.E.; Simon, M.D.; Aghia, G.; Butler, J.D. Polycomb proteins remain bound to chromatin and DNA during DNA replication in vitro. Cell 2009, 137, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Breiling, A.; O’Neill, L.P.; D’Eliseo, D.; Turner, B.M.; Orlando, V. Epigenome changes in active and inactive polycomb-group-controlled regions. EMBO Rep. 2004, 5, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Tavares, L.; Dimitrova, E.; Oxley, D.; Webster, J.; Poot, R.; Demmers, J.; Bezstarosti, K.; Taylor, S.; Ura, H.; Koide, H.; et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell 2012, 148, 664–678. [Google Scholar] [CrossRef] [PubMed]
- Vissers, J.H.; Nicassio, F.; van Lohuizen, M.; Di Fiore, P.P.; Citterio, E. The many faces of ubiquitinated histone H2A: Insights from the dubs. Cell Div. 2008, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Niessen, H.E.; Demmers, J.A.; Voncken, J.W. Talking to chromatin: Post-translational modulation of polycomb group function. Epigenetics Chromatin 2009, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; O’Loghlen, A. PRC1 complex diversity: Where is it taking us? Trends Cell Biol. 2014, 24, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Cavalli, G. Recruitment of polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development 2009, 136, 3531–3542. [Google Scholar] [CrossRef] [PubMed]
- Azuara, V.; Perry, P.; Sauer, S.; Spivakov, M.; Jorgensen, H.F.; John, R.M.; Gouti, M.; Casanova, M.; Warnes, G.; Merkenschlager, M.; et al. Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 2006, 8, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Zuscik, M.J. Regulation of chondrogenesis and chondrocyte differentiation by stress. J. Clin. Investig. 2008, 118, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Hino, S.; Murakami, T.; Kanemoto, S.; Kondo, S.; Saitoh, M.; Nishimura, R.; Yoneda, T.; Furuichi, T.; Ikegawa, S.; et al. Regulation of endoplasmic reticulum stress response by a BBF2H7-mediated Sec23a pathway is essential for chondrogenesis. Nat. Cell Biol. 2009, 11, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Arteaga-Solis, E.; McKee, M.D.; de Pablo, R.; Al Awqati, Q.; Ballabio, A.; Karsenty, G. Proteoglycan desulfation determines the efficiency of chondrocyte autophagy and the extent of FGF signaling during endochondral ossification. Genes Dev. 2008, 22, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Denoyelle, C.; Abou-Rjaily, G.; Bezrookove, V.; Verhaegen, M.; Johnson, T.M.; Fullen, D.R.; Pointer, J.N.; Gruber, S.B.; Su, L.D.; Nikiforov, M.A.; et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat. Cell Biol. 2006, 8, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Kilbey, A.; Terry, A.; Cameron, E.R.; Neil, J.C. Oncogene-induced senescence: An essential role for Runx. Cell Cycle 2008, 7, 2333–2340. [Google Scholar] [PubMed]
- Yoshida, C.A.; Komori, T. Role of Runx proteins in chondrogenesis. Crit. Rev. Eukaryot. Gene Expr. 2005, 15, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Hoenerhoff, M.J.; Bommi, P.; Sainger, R.; Guo, W.-J.; Dimri, M.; Band, H.; Band, V.; Green, J.E.; Dimri, G.P. Bmi-1 cooperates with H-Ras to transform human mammary epithelial cells via dysregulation of multiple growth-regulatory pathways. Cancer Res. 2007, 67, 10286–10295. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Jenner, R.G.; Boyer, L.A.; Guenther, M.G.; Levine, S.S.; Kumar, R.M.; Chevalier, B.; Johnstone, S.E.; Cole, M.F.; Isono, K.; et al. Control of developmental regulators by polycomb in human embryonic stem cells. Cell 2006, 125, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M.K.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Dietrich, N.; Pasini, D.; Hansen, K.H.; Helin, K. Genome-wide mapping of polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006, 20, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Chao, C.; Saito, S.; Mazur, S.J.; Murphy, M.E.; Appella, E.; Xu, Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol. 2005, 7, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, J.P.; Land, H. Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990, 18, 3587–3596. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, T.M.; Nolan, G.P. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum.Gene Ther. 1996, 7, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Brummelkamp, T.R.; Bernards, R.; Agami, R. A system for stable expression of short interferingRNAs in mammalian cells. Science 2002, 296, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Löbrich, M.; Jeggo, P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Yamaki, M.; Isono, K.; Takada, Y.; Abe, K.; Akasaka, T.; Tanzawa, H.; Koseki, H. The mouse Edr2 (Mph2) gene has two forms of mRNA encoding 90- and 36-kDa polypeptides. Gene 2002, 288, 103–110. [Google Scholar] [CrossRef]
- Hamer, K.M.; Sewalt, R.G.; den Blaauwen, J.L.; Hendrix, T.; Satijn, D.P.; Otte, A.P. A panel of monoclonal antibodies against human polycomb group proteins. Hybrid. Hybridomics 2002, 21, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Eijssen, L.M.; Jaillard, M.; Adriaens, M.E.; Gaj, S.; de Groot, P.J.; Muller, M.; Evelo, C.T. User-friendly solutions for microarray quality control and pre-processing on arrayanalysis.Org. Nucleic Acids Res. 2013, 41, W71–W76. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.J.; Irizarry, R.A.; Gentleman, R.; Martinez-Murillo, F.; Spencer, F. A model-based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc. 2004, 99, 909–917. [Google Scholar] [CrossRef]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef] [PubMed]
- Salomonis, N.; Hanspers, K.; Zambon, A.C.; Vranizan, K.; Lawlor, S.C.; Dahlquist, K.D.; Doniger, S.W.; Stuart, J.; Conklin, B.R.; Pico, A.R. Genmapp 2: New features and resources for pathway analysis. BMC Bioinform. 2007, 8, 217. [Google Scholar] [CrossRef] [PubMed]
- Kelder, T.; van Iersel, M.P.; Hanspers, K.; Kutmon, M.; Conklin, B.R.; Evelo, C.T.; Pico, A.R. Wikipathways: Building research communities on biological pathways. Nucleic Acids Res. 2012, 40, D1301–D1307. [Google Scholar] [CrossRef] [PubMed]
- Marks, H.; Kalkan, T.; Menafra, R.; Denissov, S.; Jones, K.; Hofemeister, H.; Nichols, J.; Kranz, A.; Stewart, A.F.; Smith, A.; et al. The transcriptional and epigenomic foundations of ground state pluripotency. Cell 2012, 149, 590–604. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spaapen, F.; Eijssen, L.M.T.; Adriaens, M.E.; Welting, T.J.; Prickaerts, P.; Salvaing, J.; Dahlmans, V.E.H.; Surtel, D.A.M.; Kruitz, F.; Kuijer, R.; et al. PRC1 Prevents Replication Stress during Chondrogenic Transit Amplification. Epigenomes 2017, 1, 22. https://doi.org/10.3390/epigenomes1030022
Spaapen F, Eijssen LMT, Adriaens ME, Welting TJ, Prickaerts P, Salvaing J, Dahlmans VEH, Surtel DAM, Kruitz F, Kuijer R, et al. PRC1 Prevents Replication Stress during Chondrogenic Transit Amplification. Epigenomes. 2017; 1(3):22. https://doi.org/10.3390/epigenomes1030022
Chicago/Turabian StyleSpaapen, Frank, Lars M. T. Eijssen, Michiel E. Adriaens, Tim J. Welting, Peggy Prickaerts, Juliette Salvaing, Vivian E. H. Dahlmans, Donald A. M. Surtel, Frans Kruitz, Roel Kuijer, and et al. 2017. "PRC1 Prevents Replication Stress during Chondrogenic Transit Amplification" Epigenomes 1, no. 3: 22. https://doi.org/10.3390/epigenomes1030022
APA StyleSpaapen, F., Eijssen, L. M. T., Adriaens, M. E., Welting, T. J., Prickaerts, P., Salvaing, J., Dahlmans, V. E. H., Surtel, D. A. M., Kruitz, F., Kuijer, R., Takihara, Y., Marks, H., Stunnenberg, H. G., Wouters, B. G., Vidal, M., & Voncken, J. W. (2017). PRC1 Prevents Replication Stress during Chondrogenic Transit Amplification. Epigenomes, 1(3), 22. https://doi.org/10.3390/epigenomes1030022