
Investigating the Thermal Conductance of the Cu/Si Interface Using the Molecular Dynamics Method


Abstract
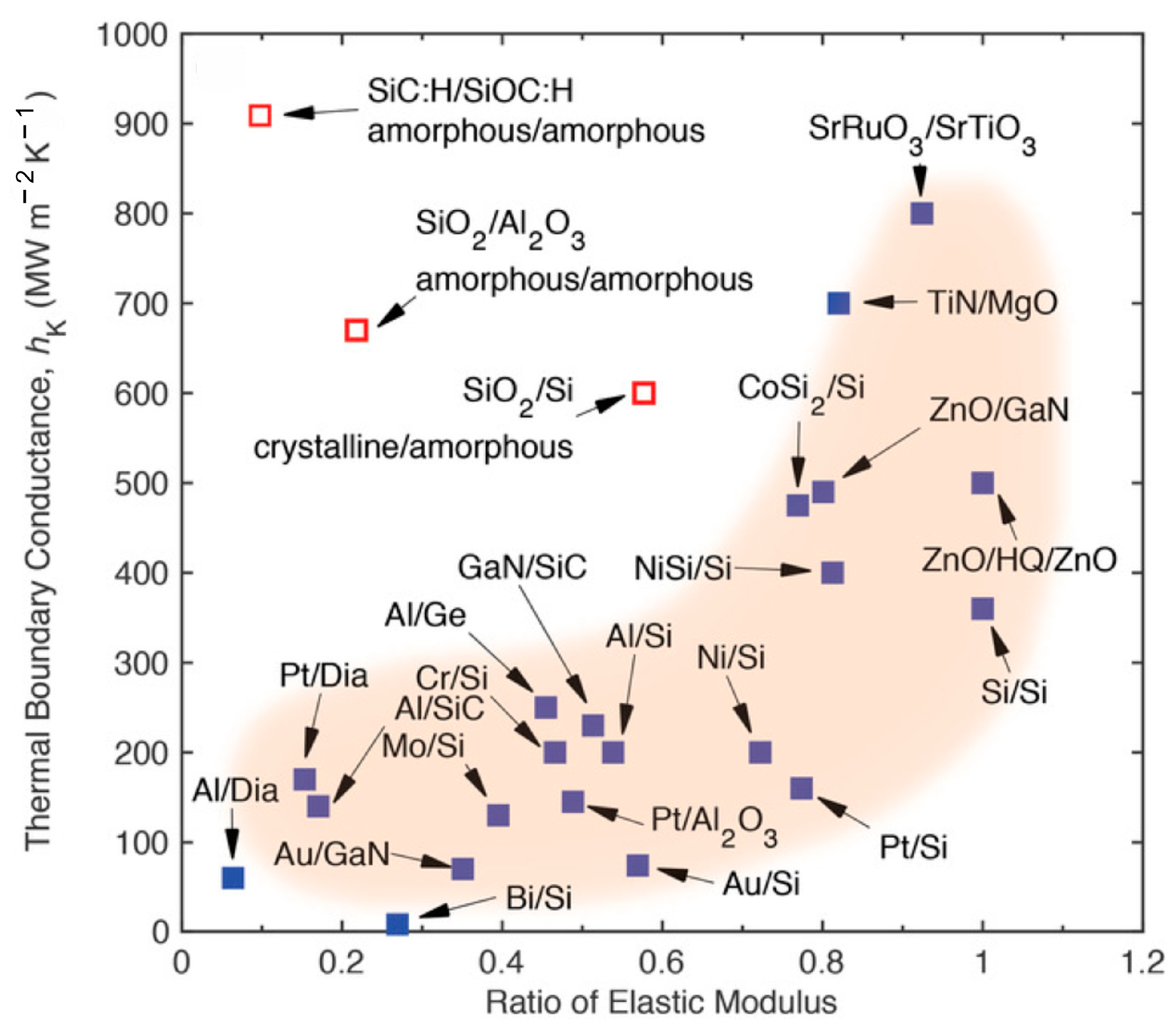
:1. Introduction
2. Calculation Methods
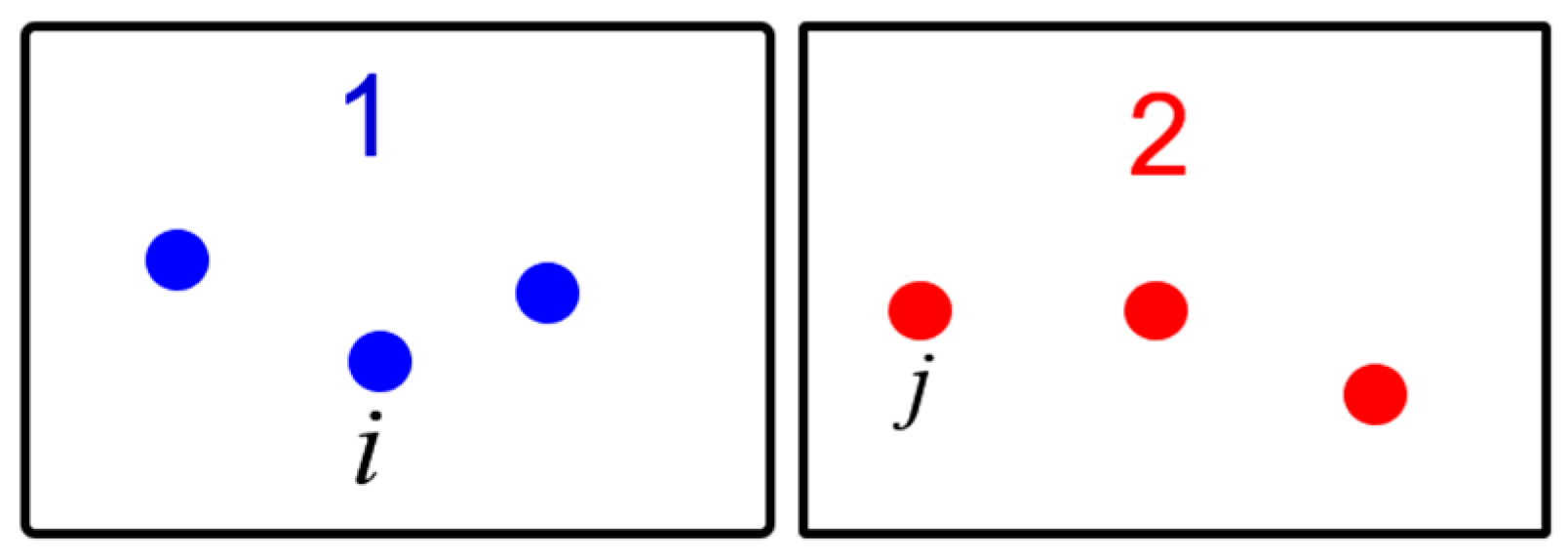
2.1. Lattice Inversion
2.2. Interface Thermal Conductance
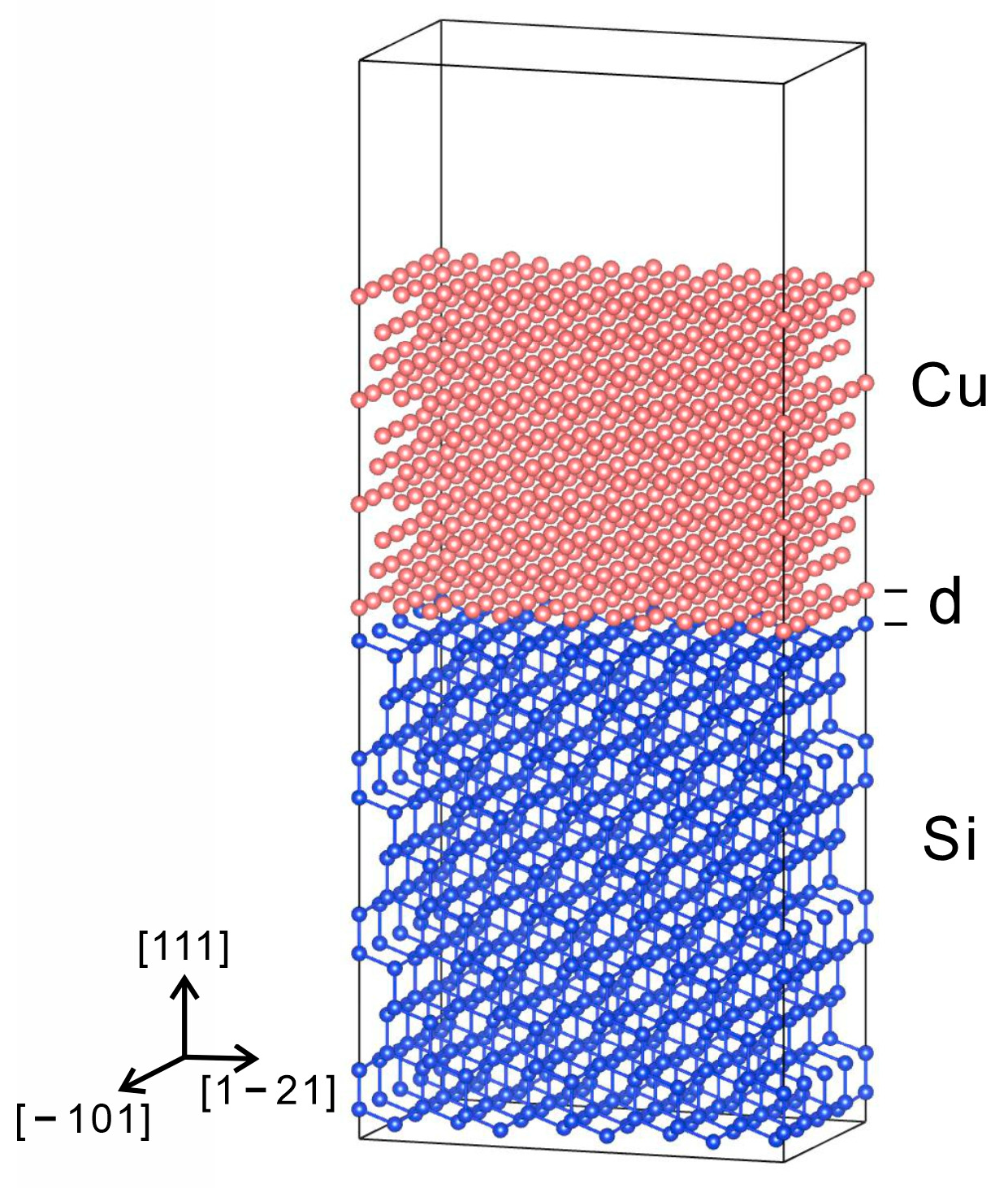
2.3. Computational Details
3. Results and Discussions
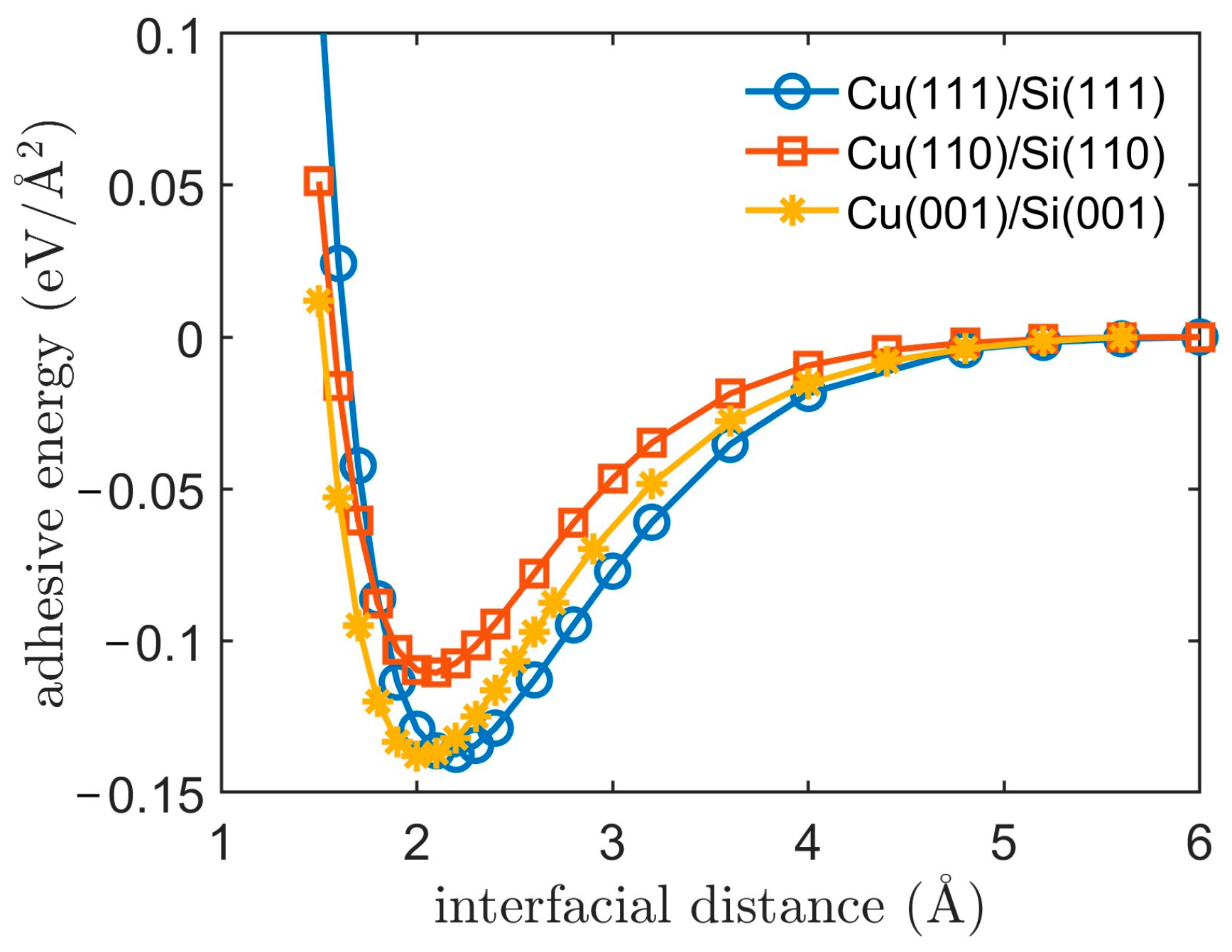
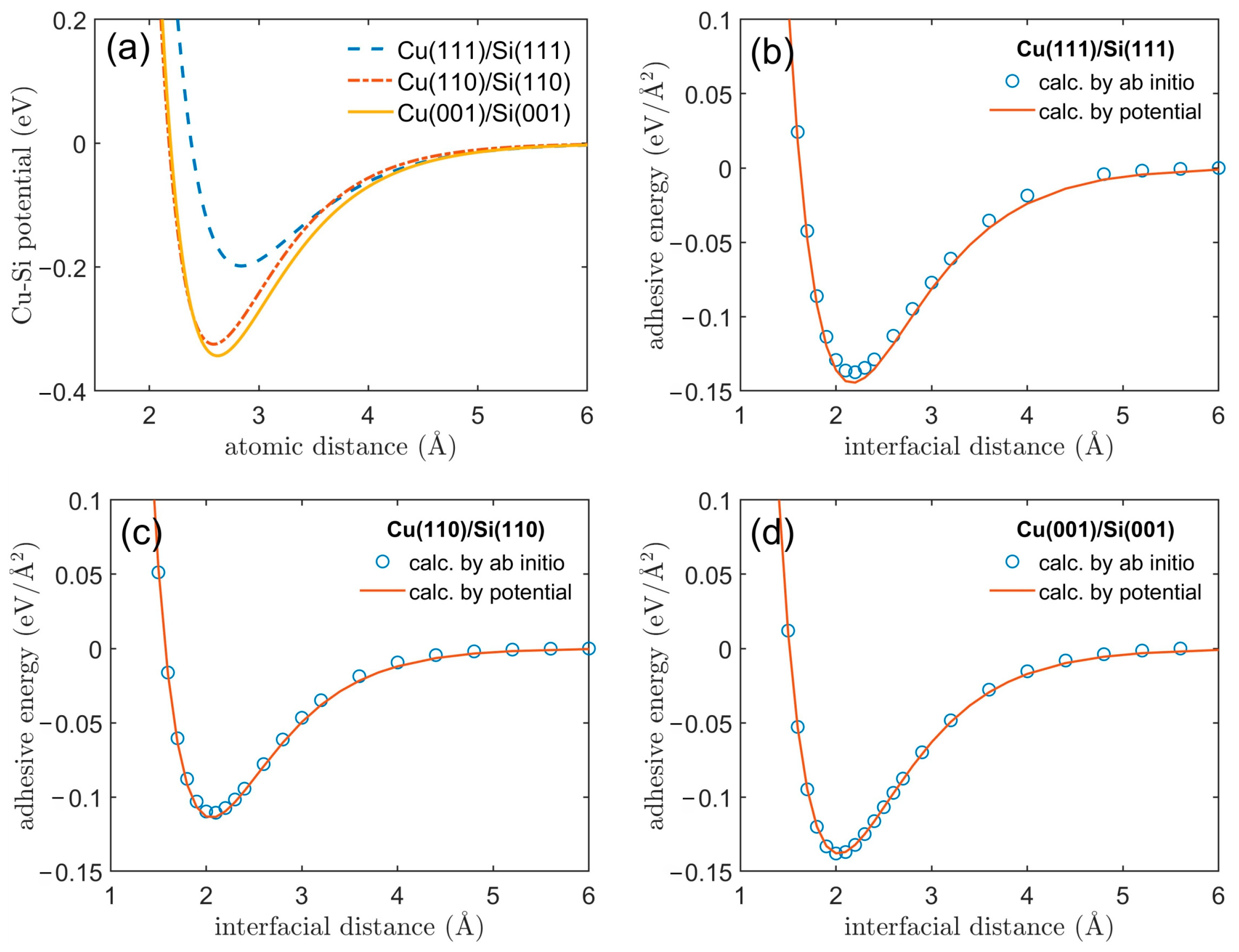
3.1. Adhesive Energy

3.2. Cu/Si Potential

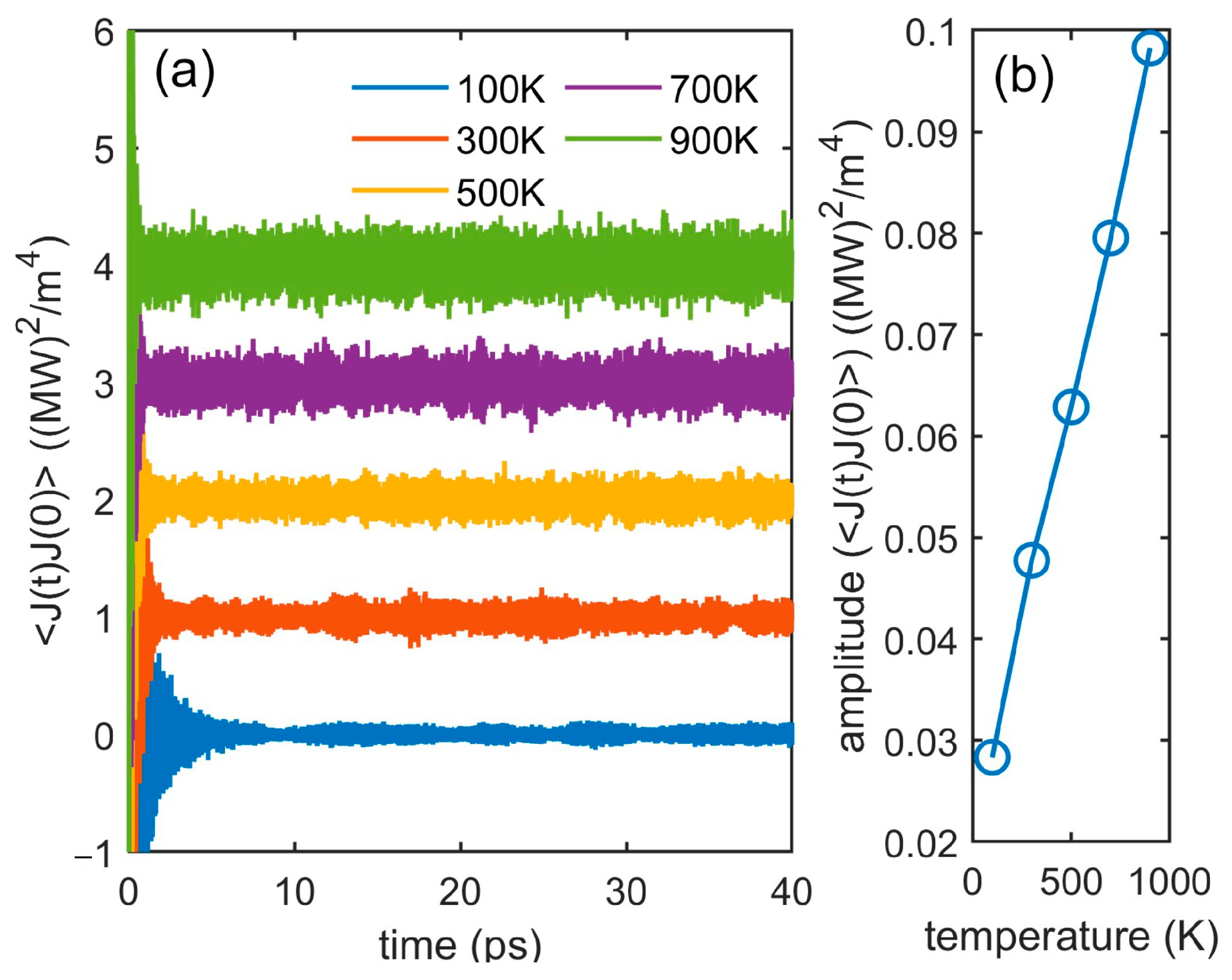
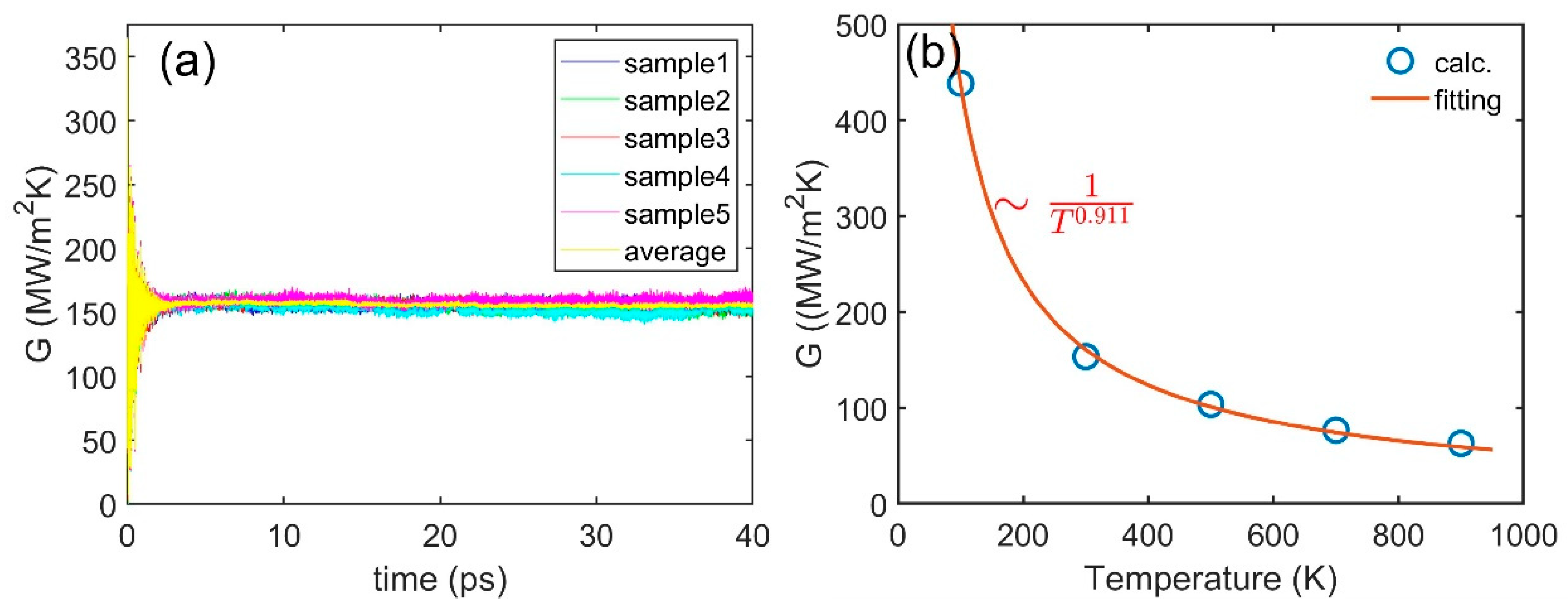
3.3. Interface Thermal Conductance
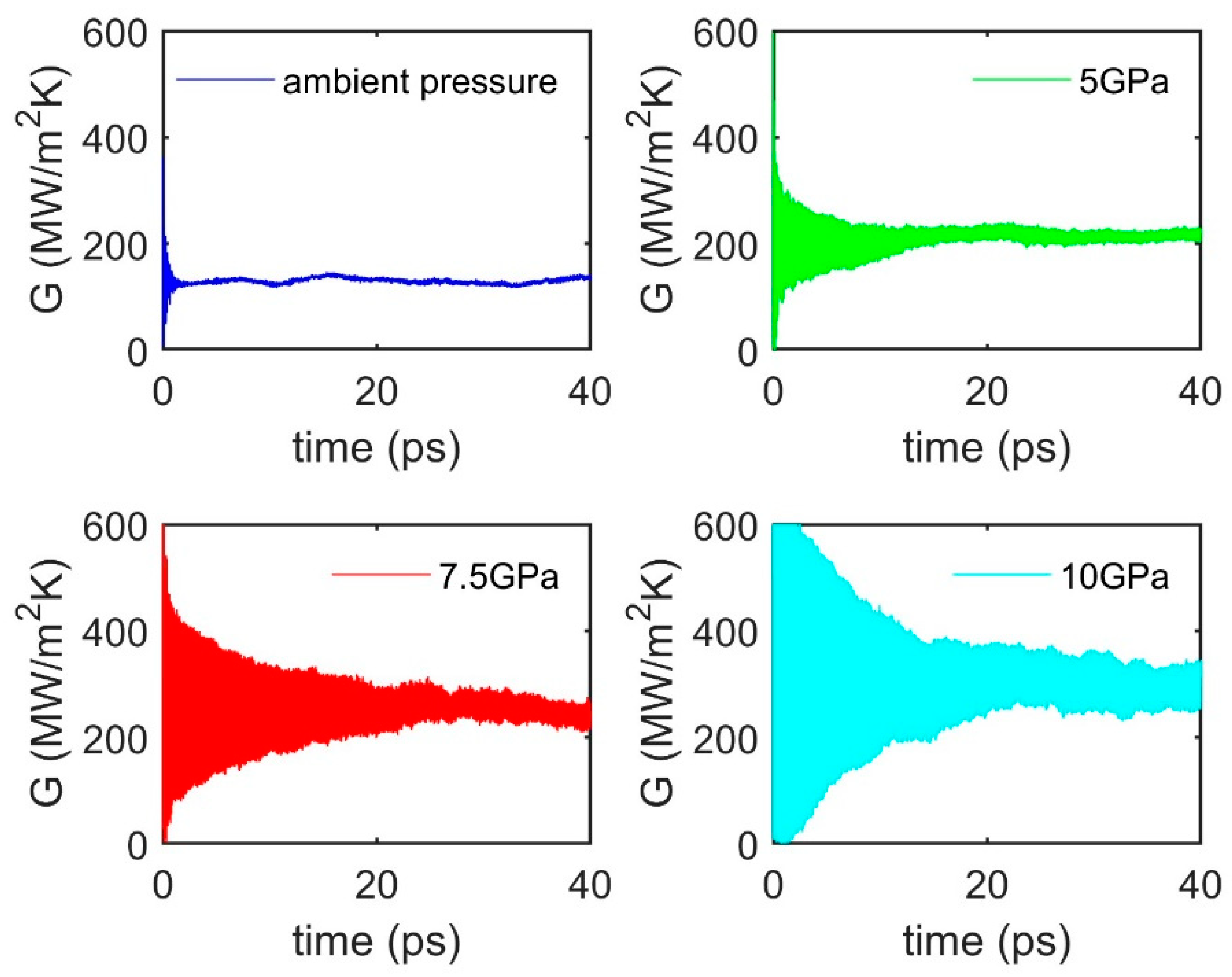
3.4. Pressure Effect
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, W.; Li, T.; Tong, Y.; Li, Y.; Wang, H.; Qi, H.; Wang, K.; Wang, H. Reducing Nonradiative Losses of Air-Processed Perovskite Films via Interface Modification for Bright and Efficient Light Emitting Diodes. Adv. Funct. Mater. 2024, 34, 2311133. [Google Scholar] [CrossRef]
- Xu, L.; Zhou, L.; Yan, M.; Luo, G.; Yang, D.; Fang, Y. High-Brightness Thermally Evaporated Perovskite Light-Emitting Diodes via Dual-Interface Engineering. Opt. Mater. 2024, 150, 115223. [Google Scholar] [CrossRef]
- Lee, W.J.; Sohn, W.B.; Shin, J.C.; Han, I.K.; Kim, T.G.; Kang, J. Growth of InGaAs/InAlAs Superlattices for Strain Balanced Quantum Cascade Lasers by Molecular Beam Epitaxy. J. Cryst. Growth 2023, 614, 127233. [Google Scholar] [CrossRef]
- Knipfer, B.; Xu, S.; Kirch, J.D.; Botez, D.; Mawst, L.J. Analysis of Interface Roughness in Strained InGaAs/AlInAs Quantum Cascade Laser Structures (λ∼4.6 μm) by Atom Probe Tomography. J. Cryst. Growth 2022, 583, 126531. [Google Scholar] [CrossRef]
- Khan, A.I.; Wu, X.; Perez, C.; Won, B.; Kim, K.; Ramesh, P.; Kwon, H.; Tung, M.C.; Lee, Z.; Oh, I.-K.; et al. Unveiling the Effect of Superlattice Interfaces and Intermixing on Phase Change Memory Performance. Nano Lett. 2022, 22, 6285–6291. [Google Scholar] [CrossRef] [PubMed]
- Aryana, K.; Gaskins, J.T.; Nag, J.; Stewart, D.A.; Bai, Z.; Mukhopadhyay, S.; Read, J.C.; Olson, D.H.; Hoglund, E.R.; Howe, J.M.; et al. Interface Controlled Thermal Resistances of Ultra-Thin Chalcogenide-Based Phase Change Memory Devices. Nat. Commun. 2021, 12, 774. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Park, S.J.; Kim, G.-Y.; Choi, S.-Y.; Jin, H. Designing Efficient Spin Seebeck-Based Thermoelectric Devices via Simultaneous Optimization of Bulk and Interface Properties. Energy Environ. Sci. 2021, 14, 3480–3491. [Google Scholar] [CrossRef]
- Wu, W.; Ren, G.-K.; Chen, X.; Liu, Y.; Zhou, Z.; Song, J.; Shi, Y.; Jiang, J.-M.; Lin, Y.-H. Interfacial Advances Yielding High Efficiencies for Thermoelectric Devices. J. Mater. Chem. A 2021, 9, 3209–3230. [Google Scholar] [CrossRef]
- Kim, H.; Kwon, Y.; Lim, H.; Kim, J.; Kim, Y.; Yeo, W. Recent Advances in Wearable Sensors and Integrated Functional Devices for Virtual and Augmented Reality Applications. Adv. Funct. Mater. 2021, 31, 2005692. [Google Scholar] [CrossRef]
- Giri, A.; Hopkins, P.E. A Review of Experimental and Computational Advances in Thermal Boundary Conductance and Nanoscale Thermal Transport across Solid Interfaces. Adv. Funct. Mater. 2020, 30, 1903857. [Google Scholar] [CrossRef]
- Findik, F. Laser Cladding and Applications. Sustain. Eng. Innov. 2023, 5, 1–14. [Google Scholar] [CrossRef]
- Chen, J. Interfacial Thermal Resistance: Past, Present, and Future. Rev. Mod. Phys. 2022, 94, 25002. [Google Scholar] [CrossRef]
- Swartz, E.T.; Pohl, R.O. Thermal Boundary Resistance. Rev. Mod. Phys. 1989, 61, 605–668. [Google Scholar] [CrossRef]
- Liang, Z.; Hu, M. Tutorial: Determination of Thermal Boundary Resistance by Molecular Dynamics Simulations. J. Appl. Phys. 2018, 123, 191101. [Google Scholar] [CrossRef]
- Little, W.A. The transport of heat between dissimilar solids at low temperatures. Can. J. Phys. 1959, 37, 334–349. [Google Scholar] [CrossRef]
- Li, Q.; Liu, F.; Hu, S.; Song, H.; Yang, S.; Jiang, H.; Wang, T.; Koh, Y.K.; Zhao, C.; Kang, F.; et al. Inelastic Phonon Transport across Atomically Sharp Metal/Semiconductor Interfaces. Nat. Commun. 2022, 13, 4901. [Google Scholar] [CrossRef] [PubMed]
- Cheaito, R.; Gaskins, J.T.; Caplan, M.E.; Donovan, B.F.; Foley, B.M.; Giri, A.; Duda, J.C.; Szwejkowski, C.J.; Constantin, C.; Brown-Shaklee, H.J.; et al. Thermal Boundary Conductance Accumulation and Interfacial Phonon Transmission: Measurements and Theory. Phys. Rev. B—Condens. Matter Mater. Phys. 2015, 91, 035432. [Google Scholar] [CrossRef]
- Stevens, R.J.; Smith, A.N.; Norris, P.M. Measurement of Thermal Boundary Conductance of a Series of Metal-Dielectric Interfaces by the Transient Thermoreflectance Technique. J. Heat Transfer 2005, 127, 315–322. [Google Scholar] [CrossRef]
- Lu, Z.; Wang, Y.; Ruan, X. Metal/Dielectric Thermal Interfacial Transport Considering Cross-Interface Electron-Phonon Coupling: Theory, Two-Temperature Molecular Dynamics, and Thermal Circuit. Phys. Rev. B 2016, 93, 064302. [Google Scholar] [CrossRef]
- Abs da Cruz, C.; Chantrenne, P.; Gomes de Aguiar Veiga, R.; Perez, M.; Kleber, X. Modified Embedded-Atom Method Interatomic Potential and Interfacial Thermal Conductance of Si-Cu Systems: A Molecular Dynamics Study. J. Appl. Phys. 2013, 113, 023710. [Google Scholar] [CrossRef]
- Chen, N. Modified Möbius Inverse Formula and Its Applications in Physics. Phys. Rev. Lett. 1990, 64, 1193–1195. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Chen, N.X.; Zhang, W.Q. Pair Potentials for a Metal–Ceramic Interface by Inversion of Adhesive Energy. J. Phys. Condens. Matter 2005, 17, 2045–2058. [Google Scholar] [CrossRef]
- Long, Y.; Chen, N.X. Pair Potential Approach for Metal/Al2O3 Interface. J. Phys. Condens. Matter 2007, 19, 196216. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, N.; Long, Y. Interfacial Potentials for Al/SiC(111). J. Phys. Condens. Matter 2009, 21, 225002. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Q.; Song, H.-Q.; Shen, J. Interfacial Potential Approach for Ag/Si(111) Interface. Mod. Phys. Lett. B 2016, 30, 1650104. [Google Scholar] [CrossRef]
- Domingues, G.; Volz, S.; Joulain, K.; Greffet, J. Heat Transfer between Two Nanoparticles Through Near Field Interaction. Phys. Rev. Lett. 2005, 94, 085901. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A Flexible Simulation Tool for Particle-Based Materials Modeling at the Atomic, Meso, and Continuum Scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Johnson, R.A. Alloy Models with the Embedded-Atom Method. Phys. Rev. B 1989, 39, 12554–12559. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.W.; Johnson, R.A.; Wadley, H.N.G. Misfit-Energy-Increasing Dislocations in Vapor-Deposited CoFe/NiFe Multilayers. Phys. Rev. B 2004, 69, 144113. [Google Scholar] [CrossRef]
- Tersoff, J. Modeling Solid-State Chemistry: Interatomic Potentials for Multicomponent Systems. Phys. Rev. B 1989, 39, 5566–5568. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interface Model | D0 (eV) | α | r0 (Å) |
|---|---|---|---|
| Cu(111)/Si(111) | 0.1983 | 1.5188 | 2.8395 |
| Cu(110)/Si(110) | 0.3247 | 1.7009 | 2.5873 |
| Cu(001)/Si(001) | 0.3434 | 1.6154 | 2.6225 |
| Cu/Si [19] | 0.9 | 1.11 | 3.15 |
| Interface Model | Temperature (K) | Pressure (GPa) | G (MW/m2K) |
|---|---|---|---|
| Cu/Si [19] | 300 | - | 436 |
| Cu/Si [20] | 300 | - | 234–263 |
| Cu(110)/Si(110) | 300 | 0 | 113 |
| Cu(001)/Si(001) | 300 | 0 | 192 |
| Cu(111)/Si(111) | 100 | 0 | 438 |
| 300 | 0 | 153 | |
| 500 | 0 | 101 | |
| 700 | 0 | 74 | |
| 900 | 0 | 59 | |
| 300 | Ambient | 125 | |
| 300 | 5 | 213 | |
| 300 | 7.5 | 248 | |
| 300 | 10 | 290 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Zhi, Y.; Song, H.; Li, H.; Wang, W.; Hu, X.; Zhang, D. Investigating the Thermal Conductance of the Cu/Si Interface Using the Molecular Dynamics Method. Metals 2024, 14, 453. https://doi.org/10.3390/met14040453
Liu S, Zhi Y, Song H, Li H, Wang W, Hu X, Zhang D. Investigating the Thermal Conductance of the Cu/Si Interface Using the Molecular Dynamics Method. Metals. 2024; 14(4):453. https://doi.org/10.3390/met14040453
Chicago/Turabian StyleLiu, Shuai, Yueyi Zhi, Hongquan Song, Huijin Li, Weiping Wang, Xiaoyan Hu, and Dongbo Zhang. 2024. "Investigating the Thermal Conductance of the Cu/Si Interface Using the Molecular Dynamics Method" Metals 14, no. 4: 453. https://doi.org/10.3390/met14040453
APA StyleLiu, S., Zhi, Y., Song, H., Li, H., Wang, W., Hu, X., & Zhang, D. (2024). Investigating the Thermal Conductance of the Cu/Si Interface Using the Molecular Dynamics Method. Metals, 14(4), 453. https://doi.org/10.3390/met14040453